


Abstract
We have investigated several in silico and in vitro methods to improve our ability to predict potential drug interactions of antibiotics. Our focus was to identify those antibiotics that activate pregnane X receptor (PXR) and induce CYP3A4 in human hepatocytes and intestinal cells. Human PXR activation was screened using reporter assays in HepG2 cells, kinetic measurements of PXR activation were made in DPX-2 cells, and induction of CYP3A4 expression and activity was verified by quantitative polymerase chain reaction, immunoblotting, and testosterone 6β-hydroxylation in primary human hepatocytes and LS180 cells. We found that in HepG2 cells CYP3A4 transcription was activated strongly (>10-fold) by rifampin and troleandomycin; moderately (≥7-fold) by dicloxacillin, tetracycline, clindamycin, griseofulvin, and (≥4-fold) erythromycin; and weakly (>2.4-fold) by nafcillin, cefaclor, sulfisoxazole, and (>2-fold) cefadroxil and penicillin V. Similar although not identical results were obtained in DPX-2 cells. CYP3A4 mRNA and protein expression were induced by these antibiotics to differing extents in both liver and intestinal cells. CYP3A4 activity was significantly increased by rifampin (9.7-fold), nafcillin and dicloxacillin (5.9-fold), and weakly induced (2-fold) by tetracycline, sufisoxazole, troleandomycin, and clindamycin. Multiple pharmacophore models and docking indicated a good fit for dicloxacillin and nafcillin in PXR. These results suggest that in vitro and in silico methods can help to prioritize and identify antibiotics that are most likely to reduce exposures of medications (such as oral contraceptive agents) which interact with enzymes and transporters regulated by PXR. In summary, nafcillin, dicloxacillin, cephradine, tetracycline, sulfixoxazole, erythromycin, clindamycin, and griseofulvin exhibit a clear propensity to induce CYP3A4 and warrant further clinical investigation.
The cytochromes P450 are responsible for the metabolism of >90% of existing pharmaceuticals, including >50% of therapeutically important drugs (Williams et al., 2004; de Groot, 2006). Clinically relevant adverse drug reactions occur due to consumption of multiple xenobiotics that interact with CYP3A4 as substrates, inhibitors, or inducers (Yao and Levy, 2002; Marechal et al., 2006; Harmsen et al., 2007). CYP3A4 inducers can cause autoinduction of clearance, or enhance clearance and decrease therapeutic efficacy of coadministered medications. The nuclear hormone receptor pregnane X receptor (PXR)/steroid and xenobiotic receptor, NR1l2, mediates transcriptional induction of CYP3A4 by many xenobiotics and endobiotics (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998). Ligand activators of PXR include a growing list of structurally and pharmacologically diverse endobiotics (e.g., pregnanes, glucocorticoids, and some bile acids), vitamins E and K2, and xenobiotics including drugs (e.g., rifampin and protease inhibitors) and environmental contaminants (e.g., organochlorine pesticides and polychlorinated biphenyls) (Ekins et al., 2007). Upon ligand binding, hPXR binds its cognate response elements in the 5′-flanking region of the CYP3A4 gene to activate its transcription.
Activation of PXR is recognized as the molecular mechanism responsible for many drug interactions (Sinz et al., 2006). Accordingly, this led to the development of high-throughput cell-based assays to test whether xenobiotics would induce CYP3A4 promoter-luciferase reporter plasmids in a PXR-dependent manner (Moore and Kliewer, 2000). However, this powerful assay only screens for whether a test compound can ligand-activate PXR and induce CYP3A4 transcription in the context of the transfected host cell. It does not reveal whether activation of CYP3A4 transcription ultimately results in induction of CYP3A4 mRNA and protein in hepatocytes, nor whether CYP3A4 activity is also increased, since some inducers are also CYP3A4 inhibitors (Piscitelli and Gallicano, 2001). It should also be noted that there are species differences in CYP3A induction that could be due to differences in PXR (LeCluyse, 2001). The standard screening assay for induction of hepatic CYP3A4 protein and activity remains primary cultures of human hepatocytes (Luo et al., 2002). Since induction of CYP3A4 in the intestine contributes to some drug interactions, and since induction of PXR targets can be ligand-, promoter-, and tissue-specific (Koch et al., 2002), it is important to test for CYP3A4 induction potential in intestine with human intestinal cell lines, such LS180s.
A variety of computational models ranging from pharmacophores (Ekins and Erickson, 2002; Schuster and Langer, 2005), quantitative structure-activity relationships and ligand docking into a PXR crystal structure (Gao et al., 2007; Lemaire et al., 2007) have all been used to predict PXR ligand binding. These computational methods generally focused on diverse structures for agonists, rarely using close structural analogs (Ekins et al., 2007). Accurate predictions can be difficult due to the size and flexibility of the human PXR ligand binding domain; however, the combination of models for searching molecule databases represents a rapid way to prescreen molecules before in vitro testing, as demonstrated previously using pharmacophores for other proteins (Ekins et al., 2005a; Chang et al., 2006) as originally suggested with the first human PXR pharmacophore (Ekins and Erickson, 2002).
The prototypical human PXR ligand, rifampin, is a potent activator of human PXR (Moore et al., 2000) and causes numerous drug interactions (Finch et al., 2002). Surprisingly, the majority of antibiotics have not been tested as potential PXR ligands despite the fact that 1) antibiotics have a long history of use (Bud, 2007), 2) they represent some of the most widely prescribed medications for treating infections (Cizman, 2003; Eng et al., 2003), 3) they are frequently taken with other medications (Pai et al., 2006), and 4) there is anecdotal data that some antibiotics can increase metabolism and decrease efficacy of coadministered medications, [particularly oral contraceptives (OCs); Weaver and Glasier, 1999]. For example, nafcillin significantly lowered the levels of the CYP3A4 substrate nifedipine (Lang et al., 2003). Flucloxacillin in combination with cyclosporine caused rejection in three of the seven kidney transplant patients (Cynke et al., 1999). There are also reports that women taking OCs who begin antibiotic regimens are more likely to experience contraception failure and breakthrough bleeding (Weaver and Glasier, 1999). However, there is clearly confusion in advising patients which antibiotics are likely to cause these drug interactions, and there is a need for some evidence-based guidelines (DeRossi and Hersh, 2002). Therefore, we designed a study whose aim was to improve our ability to predict potential antibiotic interactions by identifying with in silico and in vitro methods those antibiotics that activate PXR and induce CYP3A4 in human hepatocytes and intestinal cells.
Materials and Methods
Reagents. Rifampin (RIF), penicillins [penicillin V (PNCV), nafcillin (NFC), dicloxacillin (DXC), amoxicillin (AXC), ampicillin (APC)], cephems [cefadroxil (CFDX), cephradine (CPRD), cephalexin (CPLX), cefaclor (CFCL), cefuroxime (CFXM)], tetracyclines [tetracycline (TCL), toxycycline (DXCL), minocycline (MCL), demeclocycline (DCCL)], sulfonamides [sufisoxazole (SXZ), sulfamethoxazole (SMXZ)], macrolides [erythromycin (ERM), troleandomycin (TAO)], and others [griseofulvin (GSF), clindamycin (CMC)] (Supplemental Table 1) were all purchased from Sigma-Aldrich (St. Louis, MO) and were the highest purity available. Rifampin was used in a concentration of 10 μM. All other antibiotics were in a concentration of 50 μM unless otherwise specified (see Reporter Gene Assay Using DPX-2 Cells).
Plasmids. pSG5-hPXR was generously provided by Dr. Steven Kliewer (University of Texas Southwestern Medical Center, Dallas, TX). The reporter plasmid [CYP3A4 + 53 to 362(7836/7208)-LUC], hereafter called CYP3A4PXRE2-LUC, was prepared by Dr. Rommel Tirona (Vanderbilt University, Nashville, TN) as described previously (Tirona et al., 2003).
Reporter Gene Assay Using HepG2 Cells. HepG2 cells were maintained in minimum Eagle's medium supplemented with 10% fetal bovine serum (HyClone Laboratories, Logan, UT), 1% penicillin/streptomycin, and 1% l-glutamine. Cells were plated in 24-well plates at 3 × 105 cells per well. Twenty-four hours later, they were transfected overnight by calcium phosphate precipitation with 300 ng of PXRE2-CYP3A4-LUC reporter plasmid in the presence or absence of either pSG5-hPXR (100 ng) expression plasmid or empty vector plasmid. The next day, cells were washed once with medium and incubated with fresh medium containing serum with or without antibiotics. Twenty-four hours later, cells were harvested, lysed, and centrifuged at 1500g for 5 min, and luciferase activities were determined on an aliquot of supernatant according to the manufacturer's instructions (Luciferase Assay System; Promega, Madison, WI) using an automated luminometer (model OPTO-COMP 1; MGM Instruments, Hamden, CT). LUC activities were normalized to protein concentration. All experiments were performed at least twice in triplicate.
Reporter Gene Assay Using DPX-2 Cells. The DPX-2 cells were a generous gift from Puracyp (Carlsbad, CA). We used DPX-2 cells in a reporter gene assay for PXR as we have described recently (Ekins et al., 2007). All test articles were used in the following final concentrations: 1, 5, 10, 20, and 50 μM except for rifampin, which was used throughout at the single concentration of 10 μM. Rifampin was the positive comparator, and DMSO was used as the negative control (Ekins et al., 2007).
EC50 and Emax values were also estimated as described previously (Ekins et al., 2007). Relative induction scores (RIS) were computed as described by Ripp et al. (2006), using prior published values for Cmax and fraction unbound to calculate Cmax unb.
Preparation of Primary Cultures of Human Hepatocytes and LS180 Cells. Livers were provided by the Liver Tissue Procurement and Distribution System (National Institutes of Health Contract N01-DK-9-2310) and by the Cooperative Human Tissue Network (The Ohio State University, Columbus, OH). Livers were procured from donor organs that were not suitable for whole organ transplantation or from remaining tissue after reduced allograft transplantation. Donor livers were flushed, in situ, and maintained with Belzar's UW solution (Barr Laboratories, Pomona, NY). Hepatocytes were isolated within 24 h of cross-clamp. Reasons for not using tissues for transplantation included traumatic damage, errors in organ harvest, brief anoxic periods, or macro- or microsteatosis. Human hepatocytes were isolated essentially as described previously (Strom et al., 1996). Human hepatocyte preparations 1180, 1183, 1188, and 1201 were used for the antibiotic studies, and human hepatocyte preparations 812, 818, and 819 for the screen of other PXR activators.
Cells were plated on collagen-coated six-well plates maintained in modified Williams' E medium for 48 h and then treated with drugs for 48 h. Media were then aspirated, total lysate was harvested, or TRIzol solution (Invitrogen) added for RNA isolation. First-strand cDNA was synthesized from 2 μg of total RNA according to the manufacturer's instructions (SuperScript Preamplification System for FirstStrand cDNA synthesis; Invitrogen).
LS180 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% l-glutamine. Cells were plated on six-well plates for 24 h and then treated with drugs for 48 h. Media were then aspirated, and total lysate was harvested for protein analysis.
Quantitative PCR for hCYP3A4 mRNA. Specific PCR primers of hCYP3A4 and human glyceraldehyde-3-phosphate dehydrogenase (hGAPDH) were designed with PRIMER3 (http://www.genome.wi.mit.edu/cgi-bin/primer/primer3.cgi). The sequence homology and specificity were checked by using BLASTn (http://www.ncbi.nlm.nih.gov). The sequence of these primers were as follows: hCYP3A4.F, 5′-CACAGATCCCCCTGAAATTAAGCTTA-3′; hCYP3A4.R, 5′-AAAATTCAGGCTCCACTTACGGTG-3′; hGAPDH.F, 5′-ACCACAGTCCATGCCATCAC-3′; and hGAPDH.R, 5′-TCCACCACCCTGTTGCTGTA-3′.
Real-time PCR quantitation was carried using a QuantiTect SYBR Green PCR kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions. Amplification was done with the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). The hCYP3A4 and hGAPDH primers were used in real-time PCR amplifications in which the initial activation step was conducted at 95°C for 15 min and was followed by 40 cycles in which each cycle consisted of denaturation at 92°C for 30 s, annealing at 60°C for 30 s, and synthesis at 72°C for 60 s. Specificity of amplification was confirmed in each case by performing melt curve analysis. The relative amounts of hCYP3A4 mRNAs in each human liver sample were normalized to the hGAPDH values to control for quality of the mRNA. Quantitative PCR values were determined for CYP3A4 mRNA levels using the comparative CT (DDCT) method.
Immunoblot Analysis. Total lysates were recovered from primary cultures of human hepatocytes and LS180 cells. Protein was estimated by using the Bio-Rad protein assay with bovine serum albumin as the standard. Five micrograms and 25 μg of total lysate from human hepatocytes and LS180, respectively, were separated on 10% polyacrylamide gels, and immunoblotted with monoclonal anti-CYP3A4 K03 (Schuetz et al., 1996) and anti-mouse secondary antibody coupled with peroxidase. The blot was developed with the enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ).
CYP3A4 Enzymatic Activity. The activity of CYP3A4 was measured by formation of 6β-hydroxytestosterone in intact cultured hepatocytes, as described previously (Kostrubsky et al., 1999). After a 48-h exposure to chemicals, the culture medium was replaced with fresh Williams' E medium containing 200 μM testosterone. The cells were incubated for 30 min at 37°C, and media were taken for measurement of 6β-hydroxytestosterone, and cells were scraped and pelleted for immunoblot analysis. 6β-Hydroxytestosterone in the medium was measured by high-performance liquid chromatography with the following modifications: culture medium (100 μI) was diluted with methanol [1:1 (v/v)] and injected into a LiChrospher 100 RP-18 column (4.6 × 250 mm; 5 μm) with a mobile phase of methanol/water (60:40) at a flow rate of 1.2 ml/min. The eluent was detected by its absorbance at 242 nm and quantified by comparing the absorbance to a standard curve of 6β-hydroxytestosterone prepared in Williams' E medium.
In Silico Modeling: Pharmacophores. The computational molecular modeling studies were carried out using Catalyst in Discovery Studio 2.0 (Accelrys, San Diego, CA) running on a Centrino Duo processor (Intel, Santa Clara, CA) in a Dell Latitude D630 laptop. Pharmacophore models attempt to describe the arrangement of key features that are important for biological activity and their generation has been widely described previously (Ekins et al., 2007). Previously generated pharmacophores for PXR agonists (Ekins and Erickson, 2002; Bachmann et al., 2004; Ekins et al., 2007) were used to generate predictions for antibiotics tested in this study. These pharmacophores represent 1) the original PXR pharmacophore using 12 diverse ligands (Ekins and Erickson, 2002), with EC50 data from competition binding assays or CV1 cells, which has previously been used to predict affinity of imidazoles (Bachmann et al., 2004; Ekins et al., 2007); 2) a pharmacophore using 30 steroids, with EC50 data from HepG2 cells using a reporter assay that may define a unique site within PXR (Ekins et al., 2007); and 3) a pharmacophore using 31 diverse ligands (Ekins et al., 2007), with EC50 data from HepG2 cells using a reporter assay (Sinz et al., 2006). The structures of antibiotics were sketched in ChemDraw for Excel (CambridgeSoft, Cambridge MA) and exported as sdf files. In Catalyst, the three-dimensional molecular structures were produced using up to 255 conformers with the best conformer generation method, allowing a maximum energy difference of 20 kcal/mol. Using the Ligand Pharmacophore Mapping protocol, “Best Mapping” was performed with the “rigid fitting method” and maximum omitted features = 0. The quality of the molecule mapping to the pharmacophore is determined by the fit value, with a higher fit value representative of a better fit and dependent on the proximity of the features to pharmacophore centroids and the weights assigned to each feature.
In Silico Modeling: Docking. A selection of the antibiotics were docked into the cocrystallized structure of PXR with SR12813 (PDB ID: 1NRL) (Watkins et al., 2003) using FlexX (BioSolveIT, GmbH, Sankt Augustin, Germany) (Kramer et al., 1999; Zhang et al., 2007). The FlexX program considers ligand flexibility by an incremental ligand placement technique, whereas the receptor is considered as rigid. For each ligand, 30 different docked poses were generated, and the best pose was selected based on the FlexX score. The active site was defined as the amino acid residues within 6 Å of the cocrystallized ligand.
Statistical Analysis. Differences between two groups (control versus drug treatment) were analyzed using a two-sided, two-sample t test.
Results
Activation of PXR in a Reporter Gene Assay Using HepG2 Cells. We first screened for antibiotic activation of PXR using a CYP3A4-LUC reporter assay in the human hepatoblastoma HepG2 cells. PXR was activated strongly (>10-fold) by rifampin and troleandomycin; moderately (≥7-fold) by dicloxacillin, tetracycline, clindamycin, and griseofulvin; moderately (≥4-fold) by erythromycin; and weakly (>2.4-fold) by nafcillin, cefaclor, and sulfisoxazole (Table 1).
Maximum -fold increase in PXR activation observed experimentally in HepG2 and DPX-2 cells
Activation of PXR in a Reporter Gene Assay Using DPX-2 Cells. To determine more precise PXR binding affinities for these drugs, we next turned to the DPX-2 cells because we have previously validated their utility in combining computational and experimental data to model the interactions of a series of PXR agonists and antagonists with PXR (Ekins et al., 2007). The estimated Emax and EC50 values along with experimentally observed maximum -fold induction are listed in Table 2. Rifampin (10 μM) elicited an increase in PXR activity averaging 9.4-fold; dicloxacillin, 4.5-fold; amoxicillin, 3.8-fold; cephradine, 7.4-fold; doxycycline, 2.7-fold; democlocycline, 3.1-fold; sulfisoxazole, 2.6-fold; sulfamethoxazole, 2.3-fold; troleandomycin, 10-fold; griseofulvin, 2.3-fold; and clindamycin, 9.7-fold (Table 2). Table 2 also depicts RIS values for those antibiotics exhibiting ≥2-fold induction in DPX-2 cells.
Cmax, Cmax unb, and estimated Emax, EC50, and RIS values for antibiotics exhibiting >2-fold PXR activation in DPX-2 cells
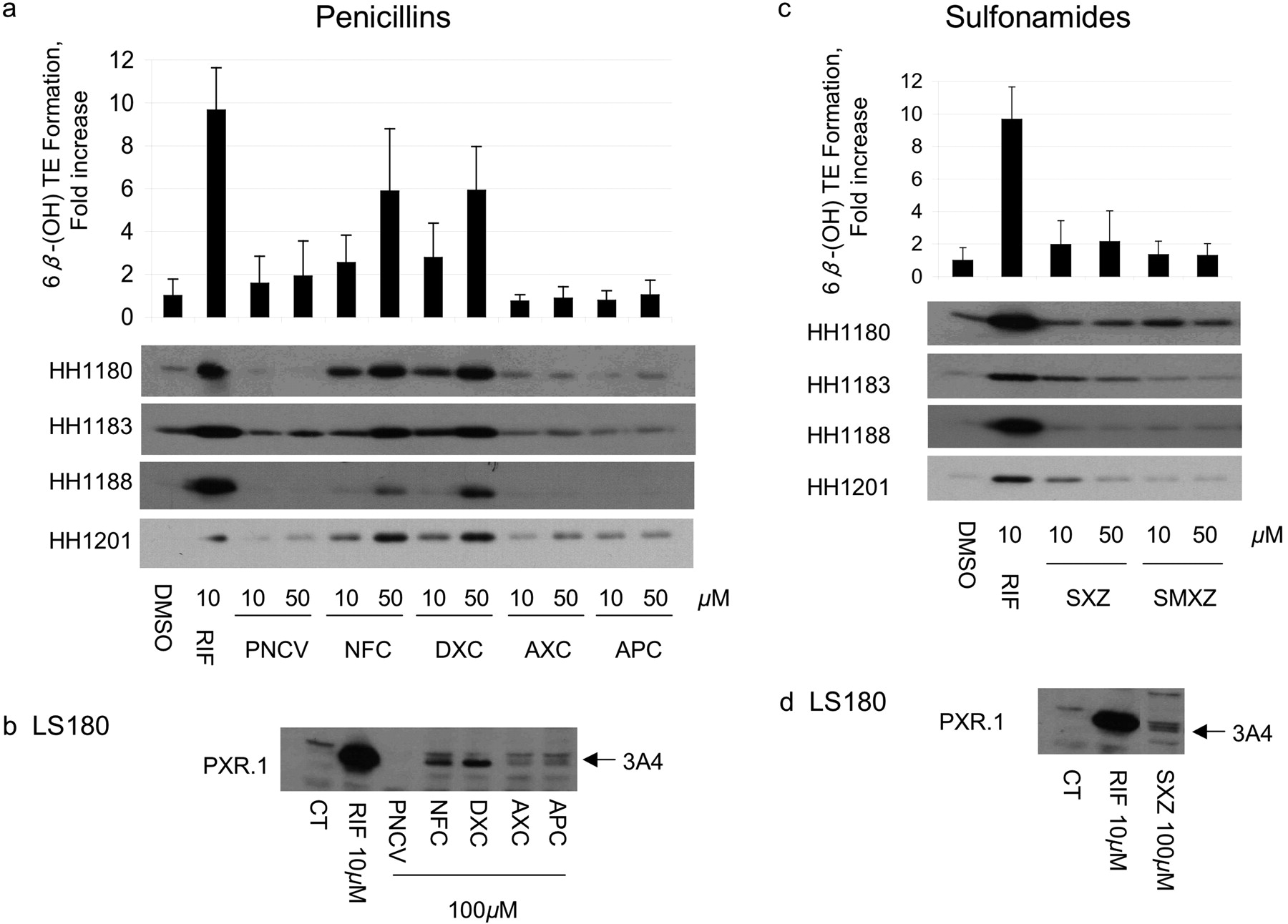
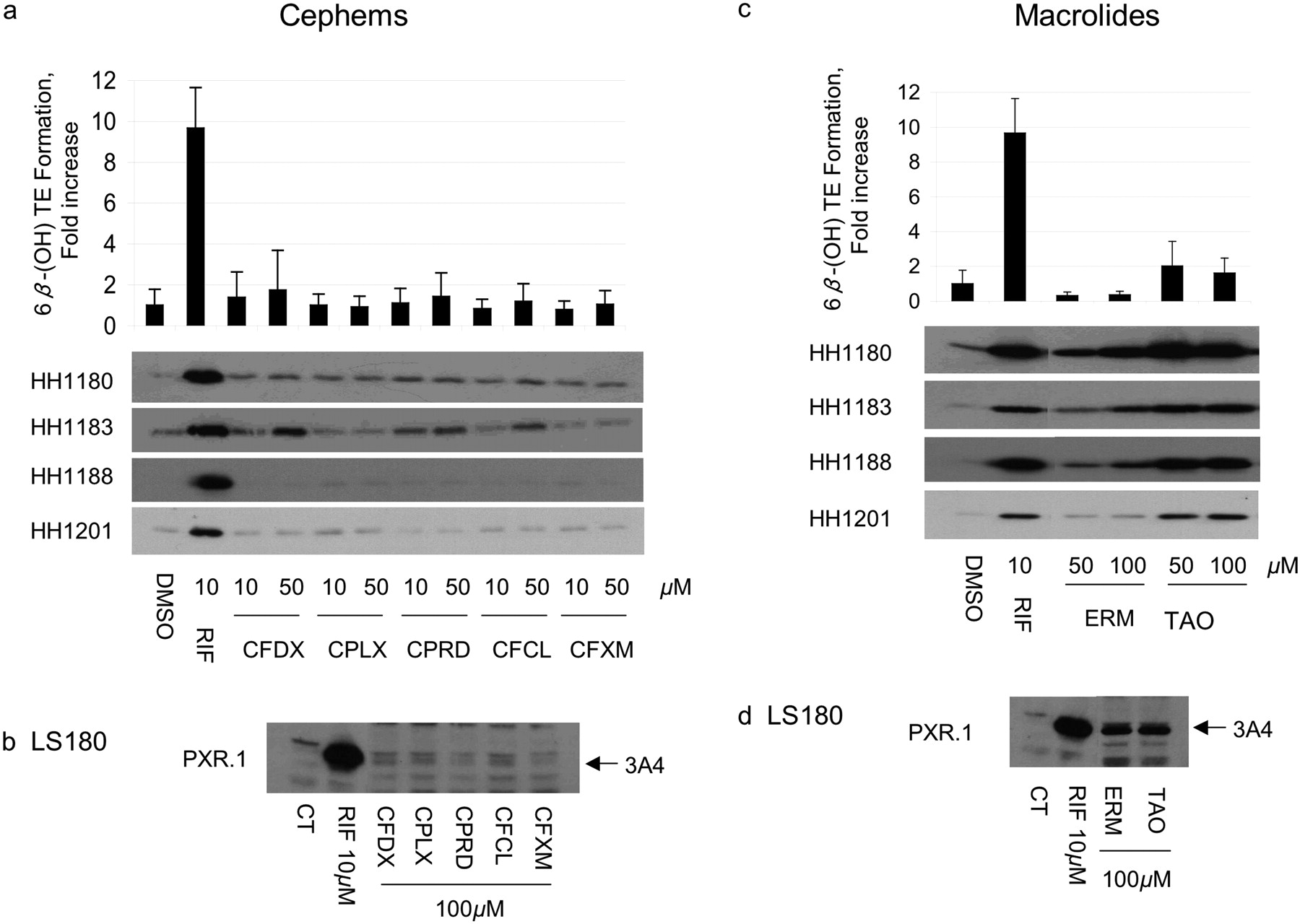
Induction of CYP3A4 mRNA, Protein, and Activity in Primary Human Hepatocytes and LS180 Human Intestinal Cells. To verify that antibiotics that transactivated PXR also resulted in increased CYP3A4, primary human hepatocytes and LS180 (human intestinal) cells were treated with the same antibiotics. CYP3A4 mRNA and protein were measured in both cell types, whereas testosterone 6β-hydroxylase activity was measured only in hepatocytes (Figs. 1, 2, 3). Among the penicillins (Fig. 1a; Table 3), nafcillin and dicloxacillin resulted in statistically significant increases in all CYP3A4 measures, approaching 6-fold induction of testosterone 6β-hydroxylase activity for both. Penicillin V resulted in an almost 2-fold increase in CYP3A4 activity. CYP3A4 protein was also induced in LS180 cells treated with these same penicillin antibiotics (Fig. 1b). Among the sulfonamides, sulfisoxazole weakly increased CYP3A4 protein in primary human hepatocytes and LS180 cells (Fig. 1, c and d), with a corresponding 2-fold increase in CYP3A4 activity (Table 3). Findings among the cephems were somewhat mixed. In primary human hepatocytes cefadroxil increased CYP3A4 mRNA and protein 1.89-fold, but these effects were not statistically significant. In contrast, although cephalexin caused a significant increase in CYP3A4 mRNA (1.67-fold), this did not result in measurable increases in CYP3A4 protein nor activity. Finally, cephradine increased CYP3A4 mRNA 2.34-fold, slightly increased CYP3A4 protein, and significantly increased CYP3A4 activity (Fig. 2, a and b; Table 3). Cefaclor caused a slight increase in CYP3A4 protein, a finding in accord with its PXR activation in HepG2 cells, although it did not seem to significantly alter any other parameter. TAO and erythromycin, which both activated PXR in HepG2 cells and DPX-2 cells (Table 1), induced CYP3A4 mRNA and protein in primary human hepatocytes and LS180 cells (Fig. 2, c and d). However, only TAO increased CYP3A4 activity (Table 3). All tetracyclines produced significant increases in hepatocyte CYP3A4mRNA; however, only tetracycline increased CYP3A4 protein. Effects on CYP3A4 activity were statistically significant for doxycycline. Both griseofulvin and clindamycin induced CYP3A4 protein in LS180 cells (Fig. 3d). However, only clindamycin caused any increase (2-fold) in CYP3A4 protein or activity in primary human hepatocytes (Fig. 3c; Table 3).
Effects of antibiotics on CYP3A4 mRNA and protein expression and CYP3A4 activity in primary human hepatocytes
There was broad, although imperfect, agreement between drugs that activated PXR in either HepG2 or DPX-2 cells and those also inducing CYP3A4 mRNA, protein, or activity. CYP3A4 mRNA, protein, or activity did not always mirror that of PXR activation. Only for rifampin, nafcillin, and dicloxacillin did the magnitude of PXR activation closely match the increases in CYP3A4 mRNA, protein, and activity.
In Silico Predictions for Antibiotics Binding to PXR as Agonists. Previously generated pharmacophores for human PXR agonists were used to predict whether antibiotics were likely to bind PXR. The original PXR pharmacophore consisted of four hydrophobes and a hydrogen bond acceptor feature and was found to map five of the antibiotics (Table 4), one of which, RIF, was originally in the pharmacophore training set. Nafcillin (Fig. 4A), dicloxacillin (Fig. 4B), erythromycin, and troleandomycin mapped to the widely dispersed features well. The diverse (n = 31) pharmacophore consisted of two hydrophobes, a hydrogen bond acceptor, and a hydrogen bond donor feature and mapped to 16 of the antibiotics, one of which, RIF, was also in the pharmacophore training set. Tetracycline, sulfisoxazole, sulfmethazole, troleandomycin, and griseofulvin did not map to the features. Interestingly, nafcillin fit well (Fig. 4C) and dicloxacillin had the lowest fit value (Fig. 4D). The steroidal (n = 30) pharmacophore consisted of four hydrophobes and a hydrogen bond acceptor feature and surprisingly was found to map four of the antibiotics: dicloxacillin (Fig. 4E), troleandomycin, clindamycin (Fig. 4F) and griseofulvin (Fig. 4G). Twelve of the antibiotics were also docked (erythromycin failed to dock) into one of the PXR crystal structures 1NRL and were then scored using FlexX (Khandelwal et al., 2007). The lower the FlexX score, the better the complimentarity between ligand and receptor. All the penicillins and cephems docked and scored well apart from cefuroxime, tetracycline, doxycycline, and clindamycin, which had poorer scores.
Using computational docking and pharmacophore methods to predict antibiotics that are likely to be PXR agonists The techniques involved in pharmacophore mapping and docking are described under Materials and Methods.
Discussion
We used HepG2 cells to conduct a primary screen, and DPX-2 cells to make precise kinetic measurements, of PXR activation and CYP3A4 transactivation by 21 antibiotics. We then verified whether these same 21 antibiotics induced CYP3A4 mRNA, protein and testosterone 6β-hydroxylase activity in primary human hepatocytes and the human intestinal LS180 cell line. However, caution is required when extrapolating PXR transactivation outcomes to predict increases in CYP3A4 activity or increases in activities associated with other known PXR target genes. Although it is likely that some other PXR target genes will be induced by the same antibiotic therapies, it is also the case that PXR-mediated gene activation is ligand-, promoter-, and cell type-specific.
There was broad, although imperfect, correlation between transactivation assays and in vitro CYP3A4 message, protein, or activity associated with cell pretreatment observed with this series of antibiotics; however, similar disparate results have also been reported for 14 more therapeutically diverse enzyme-inducing agents (Luo et al., 2002). Admittedly, the inconsistent results within and across models hampers the reliability with which in vivo outcomes can be predicted. Rifampin, our active comparator, is, of course, an exception to this problem, since rifampin markedly increased each parameter in every system. Since there are many clinical drug interaction studies with erythromycin further clinical studies would not be necessary. However, inconsistencies notwithstanding, several other antibiotics yielded sufficiently large changes in all but one or two parameters that their effects on the disposition kinetics of low clearance drugs with narrow therapeutic margins may warrant reappraisal in the context of clinical trials. Our findings suggest that nafcillin, dicloxacillin, cephradine, tetracycline, sulfisoxazole, clindamycin, and griseofulvin warrant further scrutiny in vivo in the clinic. Results for doxycycline, minocycline, demeclocycline, and sulfamethoxazole are simply too ambiguous to suggest calling for clinical reevaluation at this time. Penicillin V, amoxicillin, and ampicillin seem relatively devoid of effects on CYP3A4. Although TAO was even more active than erythromycin, its use is extremely limited in the United States.

Effect of penicillins and sulfonamides on CYP3A4 protein expression and activity in human hepatocytes and in LS180 cells. Primary human hepatocytes (HH) from donors 1180, 1183, 1188, and 1201 were treated with vehicle (DMSO), RIF, penicillins, or sulfonamides at the indicated concentrations for 48 h. HH were incubated with testosterone immediately before harvest, and the medium was analyzed for the formation of 6β-hydroxytestosterone. The -fold change [mean ± S.D. (top, a and c)] in 6β-hydroxytestosterone formation rate (nanomoles per minute per milligram of protein) for drug versus vehicle-treated cells (in triplicate) of a representative experiment is shown. The same cells were lysed, and 5 μg was analyzed by immunoblot for CYP3A4 (bottom a and c). LS180 cells stably expressing PXR.1 were treated for 48 h with the indicated concentrations of drug or 0.1% DMSO as the vehicle control (CT). Total cell lysate (25 μg) was analyzed by immunoblot analysis for CYP3A4 protein (b and d). Abbreviations for drugs are indicated in abbreviations list and Supplemental Table 1.

Effect of cephems and macrolides on CYP3A4 protein expression and activity in human hepatocytes and in LS180 cells. Primary HH from donors 1180, 1183, 1188, and 1201 were treated with vehicle (DMSO), RIF, penicillins, or sulfonamides at the indicated concentrations for 48 h. HH were incubated with testosterone immediately before harvest, and the medium was analyzed for the formation of 6β-hydroxytestosterone. The -fold change [mean ± S.D. (top, a and c)] in 6β-hydroxytestosterone formation rate (nanomoles per minute per milligram of protein) for drug versus vehicle-treated cells (in triplicate) of a representative experiment is shown. The same cells were lysed, and 5 μg was analyzed by immunoblot for CYP3A4 (bottom, a and c). LS180 cells stably expressing PXR.1 were treated for 48 h with the indicated concentrations of drug or 0.1% DMSO as the vehicle CT. Total cell lysate (25 μg) was analyzed by immunoblot analysis for CYP3A4 protein (b and d). Abbreviations for drugs are indicated in abbreviations list and Supplemental Table 1.
Antibiotics are some of the most widely prescribed drugs (Cizman, 2003; Eng et al., 2003). Their interactions with low-dose estrogen-containing OCs as a potential cause of unplanned pregnancy has been a concern among health professionals for three decades. There is considerable confusion in the marketplace as to which antibiotics may decrease the effectiveness of oral contraceptives. Back et al. (1988) reported epidemiological data showing that 63 women had become pregnant in a 17-year period in Great Britain while taking OCs, and 70% of them became pregnant while taking either penicillins or tetracyclines plus OCs. Subsequently, similar reports were described from New Zealand (Sparrow, 1989) and Holland (Kovacs et al., 1989). However, in the intervening 20 years there has been no verification that the relationship was causal. Moreover, earlier studies suggested that ampicillin had no effect on oral contraceptive effectiveness (Friedman et al., 1980; Joshi et al., 1980). Other than the rifamycins, no antibiotics have been shown to alter the pharmacokinetics of ethinylestradiol in humans (Shenfield and Griffin, 1991). Soon after implicating antibiotics as a potential cause of OC failure, Back et al., (1991) allowed that the OC failure rate among antibiotic users may not be substantively greater than the natural failure rate of OCs.
Our results show that dicloxacillin is a highly efficacious hPXR activator as well as an inducer of CYP3A4 mRNA and protein and that it can significantly increase the rate of 6β-hydroxytestosterone formation. Nafcillin is comparable for all parameters except PXR transactivation in DPX-2 cells. The findings for these two penicillins are consistent with reports of in vivo of drug interactions between these same antibiotics and CYP3A substrates. For example, Lang et al. (2003) reported that 5-day treatment with nafcillin markedly increased the total plasma clearance of the CYP3A4 substrate nifedipine by 245% in healthy controls. In a controlled trial using subjects who were infection-free, Krstenansky et al. (1987) reported that dicloxacillin decreased warfarin-induced prothrombin times (Krstenansky et al., 1987). Additional evidence that nafcillin might also increase warfarin metabolism has also been published (Cropp and Bussey, 1997).

Effect of tetracyclines, clindamycin, and griseofulvin on CYP3A4 protein expression and activity in human hepatocytes and in LS180 cells. Primary HH from donors 1180, 1183, 1188, and 1201 were treated with vehicle (DMSO), RIF, penicillins, or sulfonamides at the indicated concentrations for 48 h. HH were incubated with testosterone immediately before harvest, and the medium was analyzed for the formation of 6β-hydroxytestosterone. The -fold change [mean ± S.D. (top, a and c)] in 6β-hydroxytestosterone formation rate (nanomoles per minute per milligram of protein) for drug versus vehicle-treated cells (in triplicate) of a representative experiment are shown. The same cells were lysed, and 5 μg was analyzed by immunoblot for CYP3A4 (bottom, a and c). LS180 cells stably expressing PXR.1 were treated for 48 h with the indicated concentrations of drug or 0.1% DMSO as the vehicle CT. Total cell lysate (25 μg) was analyzed by immunoblot analysis for CYP3A4 protein (b and d). Abbreviations for drugs are indicated in abbreviations list and Supplemental Table 1.

Molecules mapped to PXR pharmacophores. Nafcillin (A) and dicloxacillin (B) mapped to the original PXR pharmacophore; nafcillin (C) and dicloxacillin (D) mapped to the diverse (n = 31) PXR pharmacophore; dicloxacillin (E), clindamycin (F), and griseofulvin (G) mapped to the steroidal (n = 30) PXR pharmacophore. Spheres represent hydrophobic features (cyan), hydrogen bond acceptor and vector (green), and hydrogen bond donor and vector (purple).
However, there are also additional reports of antibiotic drug interactions, in general with OCs. Rifampin, the most potent antibiotic activator of hPXR, was found as early as 1971 to decrease OC effectiveness. Among women taking rifampin and OCs, 75% experienced intermenstrual bleeding, and 6% became pregnant (Reimers and Jezek, 1971). The effects of rifampin on the disposition kinetics of ethinylestradiol resulting in a substantial decrease in area under the curve have been thoroughly studied (Barditch-Crovo et al., 1999). Likewise, van Dijke and Weber (1984) reported an array of case studies involving women taking concomitant griseofulvin and OCs who reported menstrual cycle disturbances and pregnancies. Based on the reported findings in humans, it is certain that rifampin can decrease the effectiveness of OCs, and it is probable that dicloxacillin and nafcillin may also behave similarly as a result of PXR activation and/or increased CYP3A4 activity. Nevertheless, a myriad of review papers and position statements caution about the potential loss of effectiveness of OCs associated with antibiotic use in general.
Our in vitro results predict that penicillin V and the aminopenicillins ampicillin and amoxicillin are unlikely to affect the metabolism of CYP3A4 substrates in humans. However, our in vitro results affirm that drug interactions are likely to occur with some other penicillins, including nafcillin and dicloxacillin, an isoxazolyl penicillin. Using LS180 cells, primary human hepatocytes, BHK21 cells, and pig kidney epithelial cells, Huwyler et al. (2006) also recently reported that clinically relevant doses of flucloxacillin (not assessed in the current study) activate PXR and induce CYP3A4 and P-glycoprotein.
Although the macrolide antibiotics erythromycin and troleandomycin were efficacious PXR activators, having both increased mRNA expression of CYP3A4, they both failed to induce CYP3A4 activity, consistent with their ability to also act as CYP3A4 inhibitors (McConn et al., 2004; Atkinson et al., 2005). Interestingly, TAO had previously been shown to have an EC50 value of 8.9 μM, whereas erythromycin was inactive in a PXR transactivation assay (Sinz et al., 2006).
Similarly, although clindamycin elicited substantial increases in PXR activation, CYP3A4mRNA, and protein, its failure to produce a significant increase in the rate of 6β-hydroxytestosterone formation could reflect its modest ability to inhibit CYP3A4 (Wynalda et al., 2003).
Computational approaches could represent a method to filter molecules before in vitro testing. Our computational analyses consisted of using multiple pharmacophores and docking into a single PXR structure. Rifampicin was present in two of the pharmacophore models; therefore, it does not merit discussion as a true prediction. However, the originally published pharmacophore based on 12 agonists (Ekins and Erickson, 2002; Bachmann et al., 2004) proved remarkably selective in only mapping nafcillin, dicloxacillin, erythromycin, and troleandomycin. The steroidal pharmacophore (with identical features) was equally selective again scoring dicloxacillin highly, along with griseofulvin and clindamycin. This was surprising because the model was derived from a set of steroidal analogs suggested to map a specific region of the PXR binding site (Ekins et al., 2007). The more structurally diverse (n = 31) pharmacophore was less discriminatory but scored nafcillin higher than other penicillins. It is interesting to note that dicloxacillin is the only antibiotic that is mapped by all three pharmacophores. A selection of the antibiotic molecules is shown to map very well to the pharmacophore features (Fig. 4). The use of multiple pharmacophores used in this way may be useful to gather a consensus prediction that could counteract the large flexible binding site, improving the overall confidence in predictions. Docking and scoring with FlexX scored the penicillins and cephems similarly and failed to dock the large erythromycin. We have also recently suggested that docking methods may need combination with other quantitative structure-activity relationships or computational methods, to improve predictions due to the flexibility of the protein and large binding site that could accommodate multiple pharmacophores (Khandelwal et al., 2007). This study represents a step in that direction.
In our transactivation studies in DPX-2 cells, a wide enough array of antibiotic concentrations was used to permit EC50 and Emax estimates that could then be used to compute a RIS (Ripp et al., 2006). By using this strategy, we identified seven of 21 (33%) antibiotics with RIS values that would predict clinically significant enzyme induction (i.e., decrease in target drug area under the curve) using the following RIS criteria (ours): likely, if an RIS score was from 0.1 to 0.5.; possible, for an RIS from 0.05 to 0.1; and not likely for an RIS <0.05. In contrast, Sinz et al. (2006) found evidence that only about 5% of a diverse array of 170 drugs that activate PXR are likely to cause clinically significant enzyme induction. It may be that some classes of drugs, such as antibiotics, show an enrichment in the number of molecules likely to show significant induction via PXR due to their possession and arrangement of key molecular features needed for interaction in the ligand binding pocket, which may be similar to those required for the therapeutic target(s). It must also be considered that antibiotics are more prone to cause drug interactions because both the doses administered and the concentrations achieved in gut and systemically are high relative to most orally administered drugs. Hence, the potential for this class of drugs to activate PXR and transactive target genes is a function not only of structure but also of antibiotic exposure.
PXR transcriptionally activates a growing list of drug detoxification genes as can be shown visually as a network of direct interactions (Ekins et al., 2005b) radiating from a central node using Ingenuity Pathways Analysis (Supplemental Fig. 1). These genes include numerous transporters and drug metabolism genes (Rosenfeld et al., 2003), leading to altered clearance of drugs that are substrates for these gene products. PXR activation by antibiotics might therefore be capable of precipitating drug-drug interactions by mechanisms other than an increase in CYP3A4 activity. We should also consider that some antibiotics such as the macrolides also inhibit P-glycoprotein (Eberl et al., 2007), and this could counteract the induction effect.
Our evaluation of 21 antibiotics using PXR transactivation in HepG2 and DPX-2 cells, CYP3A4 mRNA and protein in human hepatocytes, CYP3A4 protein in LS180 cells, and CYP3A4 activity (testosterone 6β-hydroxylase activity) in primary human hepatocytes identified a large number of antibiotics that were active in all or most of these in vitro assays for CYP3A4 induction. Rifampin, the active comparator molecule, was highly active in every assay. Dicloxacillin was also highly active in every assay, whereas nafcillin was highly active in every assay apart from transactivation in DPX-2 cells. These in vitro findings correspond well with the in silico pharmacophore predictions for the two penicillins and in vivo studies of CYP3A4 induction by these antibiotics in humans. TAO was also highly active in every assay. Erythromycin, clindamycin, and griseofulvin were also active in all assays apart from testosterone 6β-hydroxylase activity in primary human hepatoctyes, which may be an artifact of their simultaneous inhibition of CYP3A4. Although not as active as the foregoing drugs, sulfisoxazole was active in every assay, tetracycline was active in every assay except for PXR transactivation in DPX-2 cells, and cephradine was active in every assay except for PXR transactivation in HepG2 cells. Penicillin V, amoxicillin, and ampicillin were overall devoid of activity in these systems, and results with the remaining cephems, tetracyclines, and sulfamethoxazole were inconsistent and modest across these assays.
We conclude by suggesting multiple in vitro and in silico approaches are necessary to reliably predict the likelihood of clinically significant CYP3A4 induction via PXR. Among the extensive array of antibiotics that we tested, nafcillin, dicloxacillin, cephradine, tetracycline, sulfixoxazole, erythromycin, clindamycin, and griseofulvin exhibit a clear propensity to induce human CYP3A4 in vitro, and their potential to alter the disposition kinetics of drugs with narrow limits of efficacy such as ethinylestradiol in OCs warrants further investigation in the context of clinical trials.
Acknowledgments
We are grateful to Dave Baran for technical assistance with DPX-2 cells. Sean Ekins gratefully acknowledges Dr. Akash Khandelwal for providing docking scores and Dr. Cheng Chang for initial preliminary work on antibiotic predictions with PXR computational models. Ingenuity Systems Inc. graciously provided SE access to Ingenuity Pathways Analysis, and Accelrys kindly provided Discovery Studio Catalyst 2.0.
Footnotes
-
This work was supported in part by National Institutes of Health Grant GM60346, National Institutes of Health P30 CA21765 Cancer Center Support Grant, and the American Lebanese Syrian Associated Charities.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.020701.
-
ABBREVIATIONS: PXR, pregnane X receptor; h, human; OC, oral contraceptive; RIF, rifampin; PNCV, penicillin V; NFC, nafcillin; DXC, dicloxacillin; AXC, amoxicillin; APC, ampicillin; CFDX, cefadroxil; CPRD, cephradine; CPLX, cephalexin; CFCL, cefaclor; CFXM, cefuroxime; TCL, tetracycline; DXCL, doxycycline; MCL, minocycline; DCCL, demeclocycline; SXZ, sulfisoxazole; SMXZ, sulfamethoxazole; ERM, erythromycin; TAO, triacetyloleandomycin; GSF, griseofulvin; CMC, clindamycin; DMSO, dimethyl sulfoxide; RIS, relative induction score(s); PCR, polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HH, human hepatocytes; CT, control.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received January 31, 2008.
- Accepted May 19, 2008.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}