


Abstract
Our laboratory previously reported that accumulation of nonsterol isoprenoids following treatment with the squalene synthase inhibitor, squalestatin 1 (SQ1) markedly induced cytochrome P450 (CYP)2B1 mRNA and reporter activity in primary cultured rat hepatocytes, which was dependent on activation of the constitutive androstane receptor (CAR). The objective of the current study was to evaluate whether isoprenoids likewise activate murine CAR (mCAR) or one or more isoforms of human CAR (hCAR) produced by alternative splicing (SPTV, hCAR2; APYLT, hCAR3). We found that SQ1 significantly induced Cyp2b10 mRNA (∼3.5-fold) in primary hepatocytes isolated from both CAR–wild-type and humanized CAR transgenic mice, whereas the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor pravastatin had no effect. In the absence of CAR, basal Cyp2b10 mRNA levels were reduced by 28-fold and the effect of SQ1 on Cyp2b10 induction was attenuated. Cotransfection with an expression plasmid for hCAR1, but not hCAR2 or hCAR3, mediated SQ1-induced CYP2B1 and CYP2B6 reporter activation in hepatocytes isolated from CAR-knockout mice. This effect was also observed following treatment with the isoprenoid trans,trans-farnesol. The direct agonist CITCO increased interaction of hCAR1, hCAR2, and hCAR3 with steroid receptor coactivator-1. However, no significant effect on coactivator recruitment was observed with SQ1, suggesting an indirect activation mechanism. Further results from an in vitro ligand binding assay demonstrated that neither farnesol nor other isoprenoids are direct ligands for hCAR1. Collectively, our findings demonstrate that SQ1 activates CYP2B transcriptional responses through farnesol metabolism in an hCAR1-dependent manner. Further, this effect probably occurs through an indirect mechanism.
Introduction
The constitutive androstane receptor (CAR, NR1I3) is a nuclear receptor that is expressed predominately in the liver (Baes et al., 1994). Originally identified as the mediator of phenobarbital (PB)-inducible cytochrome P450 2B (CYP2B) expression (Honkakoski et al., 1998), CAR is now known to regulate a number of genes involved in the biotransformation and clearance of xenobiotics as well as endogenous compounds including bilirubin, bile acids, and thyroid hormone (Maglich et al., 2002, 2004; Huang et al., 2003). Several CAR target genes have been identified and include enzymes of phase I and II metabolism as well as drug transporters (Wang et al., 2012; Yang and Wang, 2014). More recent evidence has linked CAR to the regulation of genes involved in gluconeogenesis (Yarushkin et al., 2013; Lynch et al., 2014) and lipid metabolism (Dong et al., 2009); thus CAR is emerging as a potential target for metabolic disease.
Unlike other ligand-activated nuclear receptors, CAR exhibits substantial constitutive activity in the absence of ligand (Suino et al., 2004). In the uninduced state, CAR is localized in the cytoplasm as a multiprotein complex (Kobayashi et al., 2003; Yoshinari et al., 2003). Direct ligand binding or indirect activation stimulates CAR dephosphorylation and translocation into the nucleus (Mutoh et al., 2009), where it binds as a heterodimer with retinoid X receptor (RXR) to the regulatory regions of responsive genes (Honkakoski et al., 1996, 1998; Sueyoshi et al., 1999). Although the general structure of CAR is highly conserved, variations in the ligand-binding domain contribute to some species differences in ligand selectivity (Reschly and Krasowski, 2006; Omiecinski et al., 2011). For example, 1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) and 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) are selective ligand agonists for mouse CAR (mCAR) (Tzameli et al., 2000) and human CAR (hCAR), respectively (Maglich et al., 2003).
Several splice variants (SV) of hCAR have been identified that are suggested to contribute to functional diversity in CAR responses (Auerbach et al., 2003; Savkur et al., 2003; Jinno et al., 2004; Lamba et al., 2004; DeKeyser et al., 2011). Among the major splice variants present in the human liver, hCAR2 (SV3 or SV23) and hCAR3 (SV2 or SV24) are estimated to account for up to ∼10 and 50% of total hCAR transcripts, respectively (Jinno et al., 2004; Ross et al., 2010). Both variants encode proteins containing short insertions within the ligand binding domain (LBD); i.e., either four amino acids (SPTV) for hCAR2 or five (APYLT) for hCAR3 (Auerbach et al., 2003). In comparison with wild-type CAR (herein referred to as hCAR1), these variants exhibit low constitutive activity and appear to function as classic ligand-activated nuclear receptors, although indirect activation mechanisms have been suggested (Faucette et al., 2007; Lau and Chang, 2013). Limited information also indicates some differences in ligand selectivity among isoforms. While all variants are activated by CITCO (Auerbach et al., 2005, 2007; Lau et al., 2011), hCAR2 is also strongly activated by di-(2-ethylhexyl)phthalate (DeKeyser et al., 2009) and di-isobutyl phthalate (DeKeyser et al., 2011), whereas clotrimazole, bisphenol A, and pheniramine (Auerbach et al., 2005; Dring et al., 2010; DeKeyser et al., 2011) are agonists for hCAR3.
Regulation of hCAR by endogenous metabolites has been less well characterized. We previously reported that inhibition of the enzyme squalene synthase with squalestatin 1 (SQ1) induced CYP2B1 expression in rat liver and primary cultured rat hepatocytes (Kocarek et al., 1998). Further studies demonstrated that this effect was CAR-dependent, independent of reduced sterol synthesis, and mediated through an endogenous isoprenoid (Kocarek and Mercer-Haines, 2002). The substrate of squalene synthase, farnesyl pyrophosphate (FPP), is normally converted to squalene, leading to cholesterol biosynthesis or used to produce various nonsterol isoprenoids (Krag, 1998; Dallner and Sindelar, 2000). Squalene synthase inhibitors drive the accumulation of FPP (Jackson and Kocarek, 2008), which is dephosphorylated to farnesol (FOH) and then further metabolized to produce a family of FOH-derived dicarboxylic acids that are excreted in the urine (Bostedor et al., 1997). Treatment of primary rat hepatocytes (Kocarek and Mercer-Haines, 2002) or rats (Horn et al., 2005) with FOH also induces CYP2B1, suggesting that FOH itself might be a CAR activator, although this has not been formally demonstrated. Additionally, whether SQ1 or FOH regulate mouse or human CYP2B expression through CAR activation is not currently known. Therefore, the purpose of the current investigation was to evaluate whether SQ1 treatment activates mouse and human CAR-mediated responses by using primary cultured mouse hepatocytes. Reporter-based and coactivator recruitment assays were additionally conducted to determine the ability of SQ1 and FOH-derived metabolites to directly transactivate hCAR isoforms.
Materials and Methods
Materials.
SQ1 was a gift from GlaxoSmithKline (Research Triangle Park, NC) and pravastatin from Bristol-Myers Squibb Co. (Stamford, CT). Dimethyl sulfoxide (DMSO), CITCO, TCPOBOP, and trans,trans-FOH were purchased from MilliporeSigma (St. Louis, MO), other isoprenoids were purchased from Echelon Biosciences (Salt Lake City, UT), Matrigel from Corning Life Sciences (Tewksbury, MA), PureCol from Advanced BioMatrix (San Diego, CA), recombinant human insulin (Novolin R) from Novo Nordisk Pharmaceuticals, Inc. (Princeton, NJ), and culture medium, supplements, and Lipofectamine 2000 reagent from Invitrogen (Carlsbad, CA). Additional sources of reagents are indicated with the methods descriptions that are provided below.
Primary Cultures of Mouse Hepatocytes.
Male C57BL/6 mice CAR–wild-type (CAR-WT), C57BL/6-Nr1i3tm1.1Arte CAR-knockout mice (CAR-KO), and C57BL/6-Nr1i3tm1(NR1I3)Arte humanized CAR transgenic mice (hCAR-TG), all 5–6 weeks of age, were purchased from Taconic Farms (Germantown, New York) and maintained in an Association for Assessment and Accreditation of Laboratory Animal Care–approved animal facility with free access to chow and house-distilled water for at least 1 week prior to use. Generation of the hCAR-TG mouse model has been described previously (Scheer et al., 2008). Briefly, the hCAR (NR1I3) gene sequence from the first coding exon (exon 2) through exon 9 was knocked into the coding region of the murine Nr1i3 gene; therefore, expression of hCAR is controlled by the mouse Nr1i3 promoter. Because the entire hCAR genomic coding region is present, all potential major mRNA splice variants of human CAR (hCAR1, hCAR2, hCAR3) (Auerbach et al., 2003; Jinno et al., 2004) are expressed (Scheer et al., 2008).
Hepatocytes were isolated from mouse livers using a collagenase perfusion method, which has been described in detail elsewhere (Wu et al., 2001). Following isolation, 1.2 million hepatocytes per well were plated onto collagen I-coated six-well plates (for RNA experiments), or 0.5 million hepatocytes per well onto 12-well plates (for transfection experiments), and hepatocytes were cultured with Williams’ E medium supplemented with 0.1 μM triamcinolone acetonide, 0.25 IU/ml insulin, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Culture medium was renewed every 24 hours and, when appropriate, drugs or synthetic CAR ligands were added to the culture medium as concentrated stock solutions (1000X). Pravastatin and SQ1 were diluted in water, CITCO and TCPOBOP in DMSO, and FOH in ethanol (ETOH) at the concentrations specified in the individual figure legends. FOH (60 μM) and ETOH (0.1%) were complexed to fatty acid-free bovine serum albumin (0.05% w/v) in Williams’ E medium for at least 10 minutes prior to use.
Total RNA Isolation and Quantitative Reverse Transcription–Polymerase Chain Reaction.
Freshly isolated hepatocytes from CAR-WT, CAR-KO, or hCAR-TG mice were plated onto six-well plates, as described earlier. Beginning 24 hours after plating, culture medium was replaced with Williams’ E medium containing Matrigel (1:50 dilution) and then further incubated overnight at 37°C. The following day, cells were treated with cholesterol synthesis inhibitors or prototypical CAR activators (CITCO, TCPOBOP) at the concentrations described in the individual figure legends. Culture medium containing treatments was renewed once after 24 hours. Forty-eight hours following the initial treatment, total RNA was extracted and column purified using the PureLink RNA isolation kit (Ambion, Carlsbad, CA), and cDNA was synthesized from total RNA using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies/ThermoFisher Scientific, Grand Island, NY), each according to the manufacturer’s instructions.
Gene-specific primers to detect murine Cyp2b10, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (Hmgcr), and TATA box binding protein (product number Mm.PT.39a.22214839) mRNAs as well as the hCAR splice variants hCAR1, hCAR2, and hCAR3 were purchased from Integrated DNA Technologies (Coralville, IA). Primer sequences to detect the individual splice variants of hCAR have been published previously (Jinno et al., 2004; Zhang et al., 2013). The sequences (5′ to 3′) of primer pairs used to detect Cyp2b10 and Hmgcr are as follows: Cyp2b10: forward 5′-GCTTTGAGTACACAGACCGT-3′ and reverse 5′-CTCAAACATCTGGCTGGAGA-3′; Hmgcr: forward 5′- ATCCTGACGATAACGCGGTG-3′ and reverse 5′-AAGAGGCCAGCAATACCCAG-3′. Determination of mRNA levels was conducted using a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA) and SYBR Green Master Mix (Life Technologies/ThermoFisher Scientific). Each reaction (20 μl) contained 10 μl of SYBR green master mix, 50 ng of RNA equivalents, and 150 nM primers. The polymerase chain reaction (PCR) cycling conditions included an initial activation step (95°C, 15 minutes) followed by 40 cycles of melting (95°C, 15 seconds), and annealing/extension (60°C, 1 minute). TATA box binding protein was used as the endogenous control. All assays were performed in duplicate, and the comparative cycle threshold (ΔΔCT) method was used to calculate relative fold changes.
Generation of Plasmid Constructs.
Generation of a CAR-responsive luciferase (firefly) reporter plasmid containing the phenobarbital-responsive element (PBRE) within ∼2.4 kb of the rat CYP2B1 5′-flanking region has been described previously (Kocarek et al., 1998; Kocarek and Mercer-Haines, 2002). A CYP2B6 luciferase reporter plasmid containing both the proximal phenobarbital-responsive enhancer module (PBREM) and the distal xenobiotic-responsive enhancer module (XREM) of the human CYP2B6 gene (Wang et al., 2003) was generously provided by Dr. Thomas Chang (University of British Columbia; British Columbia, CA). Expression plasmids containing the full-length coding sequence for the major human CAR splice variants (hCAR1, hCAR2, and hCAR3) and empty vector control were gifts from Dr. Curtis Omiecinski (Pennsylvania State University, College Park, PA) (Auerbach et al., 2003).
Isoform-specific hCAR interaction with the coactivator, steroid receptor coactivator (SRC)1, was evaluated using a protein-fragment complementation assay (PCA), as described elsewhere (Remy and Michnick, 2006, 2007). Expression plasmids containing either the N-terminal region of the Gaussia princeps luciferase (Gluc-N) cDNA (corresponding to amino acids 1–93) ligated into the pEYFP-N1 expression plasmid or the C-terminal sequence (corresponding to amino acids 94–169; Gluc-C) ligated into pECFP-C1 (Clontech, Mountain View, CA) were prepared and kindly provided by Dr. James Granneman (Wayne State University, Detroit, MI). Chimeric plasmids were then constructed for the expression of fusion proteins containing either the full-length coding sequence of hCAR (isoform 1, 2, or 3) ligated downstream of Gluc-N cDNA (hCAR–Gluc-N) or the receptor interaction domain (RID) of hSRC1 ligated upstream of the Gluc-C sequence [hSRC1(RID)-Gluc-C]. The CAR and SRC1 inserts were ligated in-frame with and separated from the Gluc sequences by a polypeptide linker. Constructs were prepared by PCR using expression plasmids as templates, PfuUltraII Fusion DNA polymerase (Agilent, Santa Clara, CA), and gene-specific primers. The sequences of the primer pairs for generating all of the hCAR splice variants were: forward 5′-GCGAAGCTTTGGGCGGAGGCGGAAGCGGCGGAGGCGGAAGTAGGGAAGATGAGCTGAGGAACT-3′ (underscored sequence = HindIII site; bold = 2 nt to maintain frame; double underscored = GGGGSGGGG amino acid spacer) and reverse 5′- GCGGGATCCTCAGCTGCAGATCTCCTGGAGCA-3′ (underscored = BamHI site), corresponding to nt 210–1250 of the reference sequence for hCAR1 (NM_005122). For SRC1-RID, the forward primer sequence was 5′-GCGAAGCTTGCCACCATGGATTACAAGGATGACGACGATAAGGATGGAGACAGTAAATACTCTCAAAC-3′ (underscored sequence = HindIII site; bold = Kozak consensus sequence; italicized = start codon; double underscored= flag tag sequence), and the reverse primer sequence was 5′-GCGGGATCCAGGCTTCCGCCTCCGCCGCTTCCGCCTCCGCCGTTTGGAGTTGATCTTAAATCTTTCT-3′ (underscored sequence = BamHI; bold = 2 nt to maintain frame; double underscored = GGGGSGGGGS spacer), corresponding to nt 2119–2553 of reference sequence NM_003743. The underscored nucleotides indicate restriction sites for ligating the PCR products into the respective expression plasmid. Sequences of cloned fragments were verified by the Applied Genomics Technology Center at Wayne State University.
Transient Transfection of Primary Cultured Hepatocytes.
Primary hepatocytes isolated from CAR-KO mice were transiently transfected with CYP2B1-PBRE or CYP2B6-PBREM/XREM reporter plasmids as previously described (Kocarek and Mercer-Haines, 2002) with minor modifications. Briefly, hepatocytes were plated onto collagen I-coated 12-well plates (0.5 million hepatocytes per well), and 24 hours after plating, culture medium was replaced with fresh Williams’ E medium. For reporter assays, cells were transiently transfected with premixed complexes of Lipofectamine 2000 (4 μl), a CYP2B firefly reporter plasmid (1.5 μg), an hCAR expression plasmid or empty expression vector (50 ng), and 1 ng of the Renilla luciferase-CMV reporter plasmid (Promega Corporation, Madison, WI) diluted in 0.2 ml Opti-MEM medium. For the PCA assay, the transfection mixture contained Lipofectamine 2000 (3 μl), hCAR-Gluc-N (50 ng), and hSRC1(RID)-Gluc-C (200 ng) expression plasmids, pBluescript KS+ (1 μg), and 5 ng of the pGL3-Control firefly luciferase reporter plasmid (Promega Corporation) to allow for normalization. Culture medium was removed after 5 hours and replaced with Williams’ E medium containing Matrigel (1:50 dilution). The next day cells were treated with medium either alone or containing drug treatments as described in the individual figure legends. Culture medium containing the drug treatments was renewed after 24 hours. Hepatocytes were then harvested for measurement of luciferase activity (firefly and Renilla or firefly and Gaussia) 48 hours following initial treatment using the Dual Luciferase Reporter Assay System (Promega Corporation) and a GloMax luminometer (Promega Corporation). Treatments were performed in triplicate and repeated in two to five independent hepatocyte culture experiments (n = 3 wells/treatment per experiment).
Time-Resolved Fluorescence Resonance Energy Transfer CAR Coactivator Recruitment Assay.
FOH was diluted in either ETOH or DMSO to make a 10 mM stock concentration. Other isoprenoid stock solutions were prepared in water (FPP, isopentenyl pyrophosphate, dimethylallyl pyrophosphate, geranyl pyrophosphate, and geranylgeranyl pyrophosphate), DMSO (farnesoic acid), or ETOH (methyl farnesoate, geranylgeraniol). CITCO, prepared in DMSO, was used as a positive control hCAR agonist ligand, and clotrimazole (in DMSO) as an inverse agonist. The LanthaScreen TR-FRET CAR Coactivator Assay was used according to the manufacturer’s instructions (Invitrogen) and has been recently described (Carazo and Pavek, 2015). A dilution series of each test compound was prepared at 100× and then diluted to 2× with Complete TR-FRET Coregulator Buffer G. A 10-μl aliquot of each dilution was transferred to the wells of a 384-well plate in triplicate, and the glutathione S-transferase–hCAR ligand-binding-domain fusion protein (20 nM) was added to each well followed by a mixture containing 0.5 μM fluorescein-labeled peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC1α) peptide, and 20 nM terbium-conjugated anti-glutathione S-transferase antibody. The plate was sealed, protected from light, and incubated with gentle shaking for 1 hour at room temperature. Time-resolved fluorescence resonance energy transfer (TR-FRET) fluorescence was measured using the Victor2V Multilabel Reader (PerkinElmer, Waltham, MA) with an excitation wavelength of 340 nm and fluorescent emission signals detected at 520 (fluorescein) and 490 (terbium) nm. Emission ratios were calculated as the quotients of the emission values at 520 nm divided by the emission values at 490 nm. Binding curves were generated using the sigmoidal dose-response (variable slope) algorithm of GraphPad Prism 6 (GraphPad Software; La Jolla, CA).
Statistical Analysis.
Statistical analyses were performed using the SigmaStat statistical program (Version 3.5; Point Richmond, CA). Data for quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and transient transfections were analyzed using one-way or two-way analysis of variance as appropriate. When statistical differences were detected, the Student-Newman-Keuls test was used for individual comparisons. Results were considered significant at P < 0.05.
Results
Effect of SQ1 Treatment on Cyp2b10 and Hmgcr mRNA Levels in Primary Cultured Hepatocytes Isolated from CAR-WT and CAR-KO Mice.
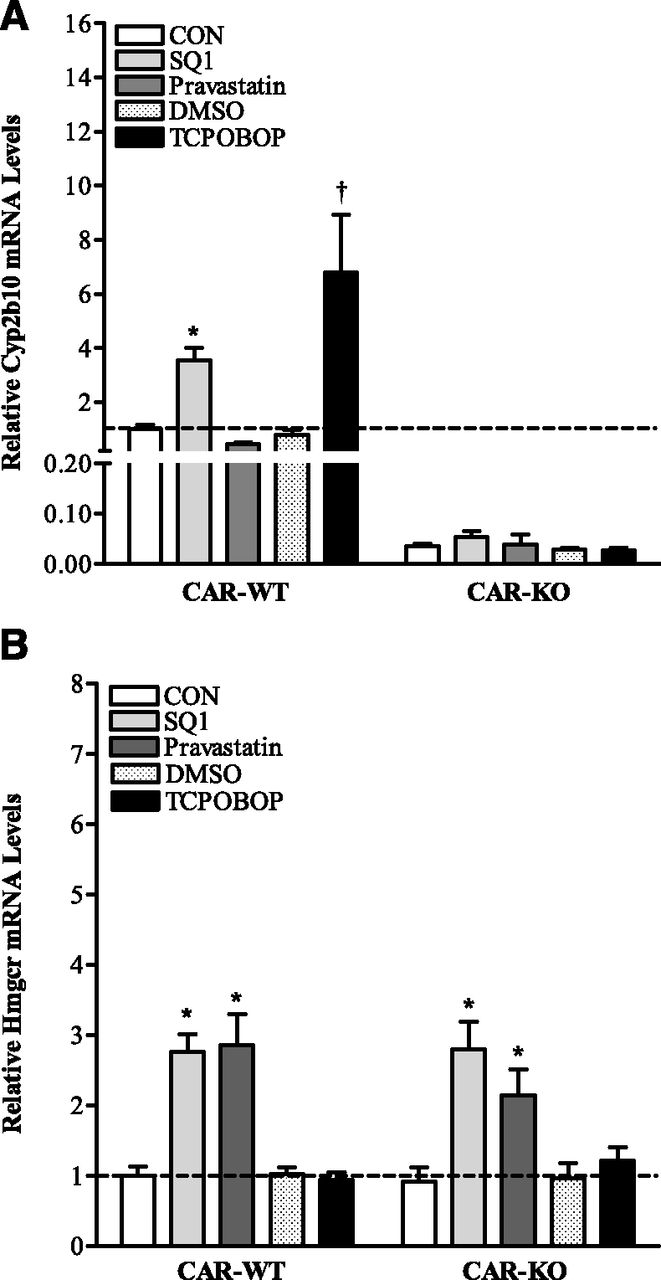
To determine whether the effect of squalene synthase inhibition on isoprenoid-mediated CYP2B expression is conserved and further investigate whether this effect is attributable to activation of one or more of the hCAR isoforms that are generated by alternative splicing, we first evaluated treatment effects on Cyp2b10 mRNA levels in primary hepatocytes isolated from CAR-WT and CAR-KO mice. As shown in Fig. 1, both SQ1 and the prototypical mouse CAR activator TCPOBOP significantly upregulated Cyp2b10 expression in a CAR-dependent manner. In hepatocytes isolated from CAR-WT mice, Cyp2b10 mRNA levels were increased ∼3.5-fold by SQ1 and 6.8-fold by TCPOBOP 48 hours after treatment (P < 0.05; Fig. 1A), whereas the HMGCR inhibitor pravastatin nonsignificantly decreased Cyp2b10 levels. In the absence of CAR, the basal level of Cyp2b10 mRNA was 28-fold lower than that measured in CAR-WT hepatocytes; however, no further significant treatment effects on Cyp2b10 levels were observed (P > 0.05; Fig. 1A).

Effect of the cholesterol synthesis inhibitors SQ1 and pravastatin on mRNA levels of (A) Cyp2b10 and (B) Hmgcr in primary cultured hepatocytes isolated from CAR-WT and CAR-KO mice. Hepatocytes were isolated from C57BL/6 (CAR-WT) and CAR-KO mice and plated onto collagen-coated six-well plates. Forty-eight hours after plating, cultures were incubated in Williams’ E medium either alone (CON) or containing 0.1% DMSO, 0.1 μM SQ1, 30 μM pravastatin, or the prototypical mouse CAR ligand agonist TCPOBOP (0.25 μM). Ninety-six hours after the initial plating, cultures were processed for the measurement of Cyp2b10 and Hmgcr mRNA levels by qRT-PCR. Values are normalized to those of the untreated (CON), CAR-WT hepatocytes. Each bar represents the mean ± S.E.M. of normalized values from 3–4 independent hepatocyte preparations. *Significantly different compared with untreated (CON) controls (P < 0.05). †Significantly different compared with DMSO controls (P < 0.05).
Depletion of intracellular cholesterol initiates transcriptional upregulation of cholesterol biosynthetic enzymes through proteolytic cleavage and activation of the sterol-sensitive transcription factors (sterol regulatory element–binding proteins, Srebps) (Sakakura et al., 2001). To demonstrate that SQ1 and pravastatin were both effective in blocking cholesterol synthesis, we evaluated Hmgcr mRNA levels in CAR-WT and CAR-KO hepatocytes (Fig. 1B). As shown in Fig. 1B, SQ1 and pravastatin, but not TCPOBOP, induced Hmgcr mRNA levels to a similar extent in both CAR-WT and CAR-KO hepatocytes (P < 0.05), consistent with sterol depletion. Because both drugs induced Hmgcr and only SQ1 induced Cyp2b10 mRNA levels, our results indicate that the effect of SQ1 on Cyp2b10 induction is independent of Srebp-mediated transcription, as previously established in rats (Kocarek and Mercer-Haines, 2002).
SQ1 Induces Cyp2b10 mRNA Levels in Primary Cultured Hepatocytes Isolated from hCAR-TG Mice.
Having demonstrated that responsiveness of Cyp2b10 to SQ1 was attenuated in CAR-KO mouse hepatocytes, we next evaluated whether SQ1 mediates Cyp2b10 induction in primary hepatocytes isolated from humanized CAR transgenic mice (hCAR-TG). In this model, expression of hCAR is controlled by the mCAR promoter; however, activation of CAR-mediated responses is hCAR-dependent (Scheer et al., 2008). Treatment effects were measured after 48 hours, and relative expression levels of the major splice variants of hCAR were also evaluated. As shown in Fig. 2, both SQ1 and the prototypical hCAR activator CITCO increased Cyp2b10 mRNA levels, although this only reached significance in SQ1-treated hepatocytes (P < 0.05), whereas pravastatin had no effect on Cyp2b10 mRNA levels. Consistent with the effects observed in wild-type mouse hepatocytes, SQ1 and pravastatin also increased Hmgcr mRNA levels to a similar extent (P < 0.05; Fig. 2B), demonstrating that both treatments were equally effective in blocking sterol synthesis and the cumulative effect of SQ1 was attributable to isoprenoid accumulation. Relative mRNA levels for the major splice variants of hCAR were also assessed (Fig. 2C). We found that among the major splice variants, hCAR1 (SV0, 59%) was most abundantly expressed, followed by hCAR3 (∼33%) and hCAR2 (∼8%), when expressed as a percentage of the total of the three isoforms. These results are consistent with levels observed in human livers and other hCAR transgenic mouse models (Jinno et al., 2004; Ross et al., 2010; Zhang et al., 2013).

Relative mRNA levels of the major human CAR splice variants (hCAR1, hCAR2, and hCAR3) and induction of Cyp2b10 mRNA expression by SQ1 treatment in primary hepatocytes isolated from hCAR-TG mice. Hepatocytes were prepared from hCAR-TG mice and plated onto collagen-coated six-well plates. Forty-eight hours after plating, cultures were incubated in Williams’ E medium either alone (CON) or containing 0.1% DMSO, 0.1 μM SQ1, 30 μM pravastatin, or the prototypical human CAR ligand agonist, CITCO (1 μM). Cultures were processed for the measurement of hCAR splice variants and Cyp2b10 and Hmgcr mRNA levels by qRT-PCR 48 hours after the initial treatment. (A) SQ1 and CITCO, but not pravastatin, induce Cyp2b10 mRNA levels in primary hepatocytes isolated from hCAR-TG mice. (B) SQ1 and pravastatin, but not CITCO, induce Hmgcr mRNA levels. (C) Levels of the major splice variants of human CAR (hCAR1, hCAR2, hCAR3) expressed relative to hCAR1 mRNA levels. Each bar represents the mean ± S.E.M. of normalized values from two mouse hepatocyte preparations (n = 2 wells/treatment per preparation). *Significant compared with untreated (CON) controls (P < 0.05). δSignificantly different compared with hCAR1 mRNA levels (P < 0.05).
SQ1 Activates CYP2B1-PBRE and CYP2B6-PBREM/XREM Reporter Activity in an hCAR1-Dependent Manner.
The previous results suggested that SQ1 treatment could activate hCAR depending on the cellular context. Therefore, we next evaluated whether the effects of SQ1 were mediated through one or more of the individual splice variants of hCAR using hepatocytes from a CAR-KO cellular background. To consider whether species differences in CAR target gene sequences could contribute to hCAR-mediated effects, luciferase reporter assays were conducted with constructs containing either the PBREM/XREM of the human CYP2B6 gene or the PBRE of the rat CYP2B1 gene. Primary hepatocytes isolated from CAR-KO mice were cotransfected with a reporter construct and each individual hCAR expression plasmid or an empty vector control, and treatment effects were evaluated after 48 hours. Results are presented in Fig. 3. For both CYP2B reporter plasmids, a significant interaction was detected between treatment and expression plasmid (P < 0.0001). In general, transfection of cells with hCAR1 significantly increased basal activity of both reporters, compared with the empty expression vector (Fig. 3, A and B; P < 0.05). Treatment of hCAR1-transfected hepatocytes with SQ1, the hCAR ligand agonist CITCO, or the indirect CAR activator PB significantly increased the activity of both reporters, compared with vehicle controls (P < 0.05; Fig. 3, A and B).
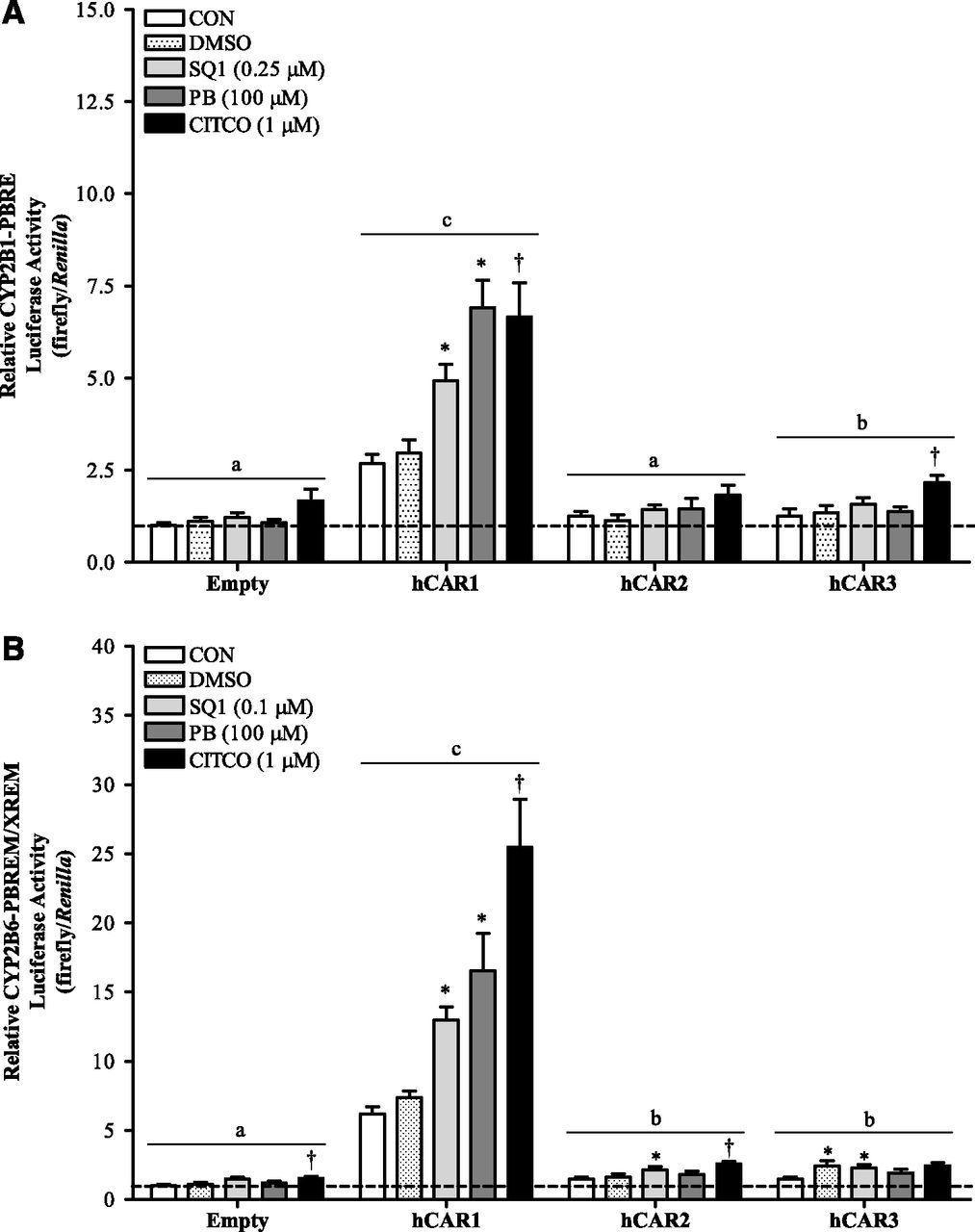
SQ1 modulates (A) CYP2B1-PBRE and (B) CYP2B6-PBREM/XREM reporter activity in an hCAR1-dependent manner. Primary hepatocytes freshly isolated from CAR-KO mice were plated onto collagen-coated 12-well plates. Twenty-four hours after plating, cultures were transfected with either a CYP2B1-PBRE or a CYP2B6-PBREM/XREM firefly luciferase reporter plasmid in combination with either an hCAR expression plasmid or empty vector, as described in Materials and Methods. Forty-eight hours after plating, cultures were incubated in Williams’ E medium either alone (CON) or containing 0.1% DMSO or SQ1 (0.1 or 0.25 μM). The prototypical human CAR activators CITCO (1 μM) and PB (100 μM) were used as positive controls. Ninety-six hours after initial plating, cultures were processed for the measurement of luciferase activity. Each bar represents the mean ± S.E.M. of normalized luciferase measurements (firefly/Renilla) combined from four to five hepatocyte preparations (n = 3 wells/treatment per hepatocyte preparation). For both reporters, a significant interaction was detected (P < 0.05). The figures show significant treatment effects within individual expression plasmids and significant differences among the different expression plasmids. *Significantly different compared with untreated controls within each expression plasmid (P < 0.05). †Significantly different compared with DMSO controls within each expression plasmid (P < 0.05). Letters denote significant effects of expression plasmid on CYP2B reporter activity; groups with different letters are significantly different (P < 0.05).
Some treatment-induced increases in luciferase activity for both reporters were evident in the empty expression vector–transfected CAR-KO hepatocytes, which were stronger for CITCO than the other treatments and somewhat variable among different hepatocyte preparations. In cells transfected with the CYP2B1 reporter, CITCO treatment significantly increased luciferase activity in the hCAR3-transfected cells (Fig. 3A). However, this CITCO-mediated increase was not significantly different from the luciferase activity measured in CITCO-treated empty vector controls (P > 0.05), indicating a weak effect of CITCO/hCAR3 on CYP2B1 activation in this system. After adjusting for background levels in the empty expression vector–transfected controls and the basal activity in vector-matched untreated controls, the adjusted fold-changes for hCAR1 were 1.5, 1.5, and 2.4; for hCAR2, 0.95, 0.87, and 1.1; and for hCAR3, 1.0, 1.0, and 1.0 in hepatocytes treated with SQ1, CITCO, and PB, respectively. In cells transfected with the CYP2B6-PBREM/XREM reporter, significant basal luciferase activity was detected with all hCAR isoforms, although this effect was weaker for the hCAR2 and hCAR3 isoforms (<2-fold) than for hCAR1 (∼6-fold) (Fig. 3B). Similar to the findings for the CYP2B1 reporter, CITCO or SQ1 treatment increased CYP2B6-PBREM/XREM reporter activity <2-fold in hCAR2 or hCAR3-transfected hepatocytes, and more strongly in hCAR1-transfected cells. After adjusting for basal activity detected among the different hCAR expression plasmids and for treatment effects within the empty vector controls, the adjusted fold-changes for hCAR1 were 1.4, 2.7, and 2.3; for hCAR2, 1.0, 1.1, and 1.1; and for hCAR3, 1.1, 1.1, and 1.1 in hepatocytes treated with SQ1, CITCO, and PB, respectively. Thus, these results indicate the main treatment effects occurred in hCAR1-transfected cells for both the CYP2B1 and CYP2B6-PBREM/XREM reporters (Fig. 3, A and B).
FOH Treatment Enhances CYP2B6-PBREM/XREM Reporter Activity in Primary Cultured CAR-KO Hepatocytes Transfected with Either hCAR1 or mCAR Expression Plasmid.
Inhibition of squalene synthase causes accumulation of the branch point intermediate FPP, which can be dephosphorylated to produce the isoprenoid alcohol FOH (Bansal and Vaidya, 1994). FOH is then oxidized to farnesoic acid and a family of dicarboxylic acids that are then excreted in the urine (Gonzalez-Pacanowska et al., 1988; Bostedor et al., 1997; Vaidya et al., 1998). Treatment of animals with squalene synthesis inhibitors or FOH produces a similar profile of metabolites in the urine, indicating that this pathway is the primary route of FPP catabolism following squalene synthase inhibition (Bostedor et al., 1997; Vaidya et al., 1998). Treatment of primary cultured rat hepatocytes or rat livers with FOH was previously shown to upregulate CYP2B1 mRNA levels (Kocarek and Mercer-Haines, 2002; Horn et al., 2005). Therefore, we additionally determined whether FOH treatment could directly influence hCAR1- or mCAR-mediated activation of the CYP2B6-PBREM/XREM reporter plasmid. As shown in Fig. 4, transfection of cells with either hCAR1 or mCAR significantly increased CYP2B6-PBREM/XREM reporter activity compared with empty vector controls (P < 0.05). Treatment of cells with FOH (60 μM) further enhanced reporter activity in the hCAR1- and mCAR-transfected cells by ∼1.5-fold compared with ETOH-treated controls (Fig. 4).

FOH potentiates hCAR1- and mCAR-mediated activation of the CYP2B6-PBREM/XREM luciferase reporter. Primary hepatocytes from CAR-KO mice were plated onto collagen-coated 12-well plates. Twenty-four hours after plating, cultures were transfected with CYP2B6-PBREM/XREM firefly luciferase reporter plasmid in combination with hCAR1 or mCAR expression plasmid or empty vector, as described in Materials and Methods. Twenty-four hours later, cultures were incubated in Williams’ E medium containing 0.05% lipid-free bovine serum albumin (BSA) together with 0.1% ETOH or 60 μM FOH. Luciferase activity was measured 48 hours after the initial treatment. Each bar represents the mean ± S.E.M. of normalized luciferase measurements (firefly/Renilla) combined from two to three independent hepatocyte preparations (n = 3 wells/treatment per hepatocyte preparation). *Significantly different compared with empty vector controls (P < 0.05). δSignificant compared with ETOH-treated hepatocytes (P < 0.05).
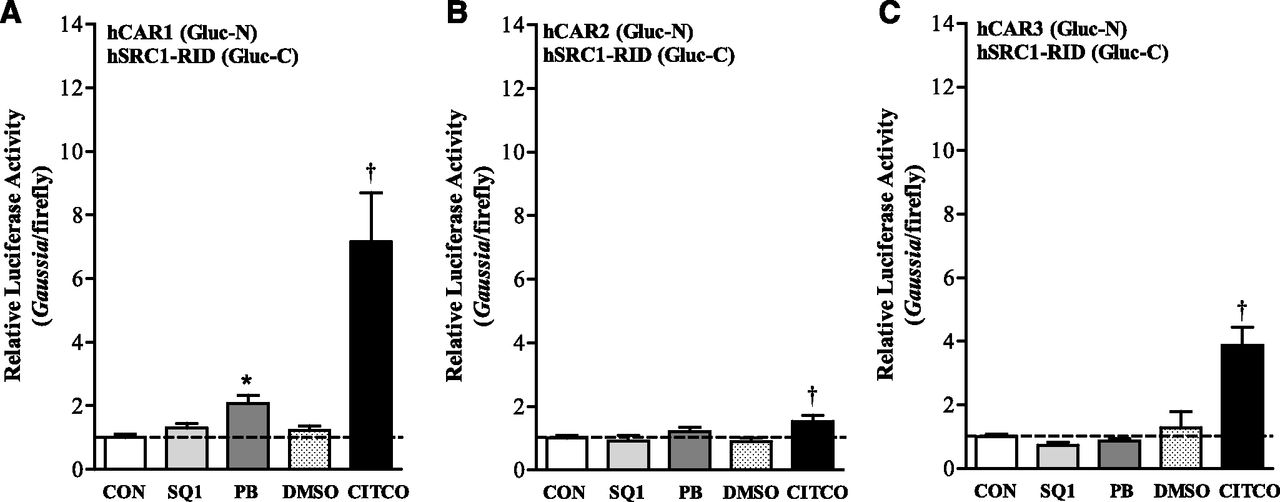
Treatment Effects of SQ1 on hCAR-SRC1 Coactivator Interaction in Primary Cultured Hepatocytes.
Unlike most ligand-activated nuclear receptors, hCAR1 can weakly interact with coactivators in the absence of a ligand, which is stabilized upon heterodimerization with RXR (Dussault et al., 2002) and even further in the presence of a ligand agonist (Wright et al., 2007). A number of coactivators have been identified that bind to CAR-WT (Muangmoonchai et al., 2001; Arnold et al., 2004; Kachaylo et al., 2011). Because all major splice variants of hCAR (hCAR1, hCAR2, and hCAR3) interact with the nuclear receptor coactivator SRC1 (Auerbach et al., 2003; Lau and Chang, 2013), we evaluated whether SQ1 treatment influenced isoform-specific hCAR-SRC1(RID) interactions using a protein-fragment complementation assay. As shown in Fig. 5, SQ1 had no significant effect on the interaction of hSRC1(RID) with any hCAR isoform, whereas the indirect CAR activator PB modestly increased hSRC1(RID) interaction with hCAR1 (P < 0.05). By comparison, the ligand agonist CITCO (1 μM) strongly increased the interaction between hSRC1(RID) and hCAR1 (∼7-fold), weakly increased the interaction with hCAR2 (1.7-fold), and moderately increased the interaction with hCAR3 (∼4-fold) (P < 0.05) (Fig. 5).

Effect of SQ1 and prototypical CAR activators on hCAR-SRC1 interaction in primary cultured hepatocytes isolated from CAR-KO mice. Primary hepatocytes freshly isolated from CAR-KO mice were plated onto collagen-coated 12-well plates. Twenty-four hours after plating, cultures were transiently transfected with chimeric plasmids for the expression of fusion proteins containing the N-terminal fragment of Gaussia luciferase fused to the entire coding sequence of (A) hCAR1 (hCAR1-Gluc-N), (B) hCAR2 (hCAR2-Gluc-N), or (C) hCAR3 (hCAR3-Gluc-N) together with an expression plasmid encoding the hSRC1 receptor interaction domain (RID) fused to the C-terminal fragment of Gaussia luciferase (hSRC1-Gluc-C), as described in Materials and Methods. Forty-eight hours following initial transfection, cultures were incubated in Williams’ E medium either alone (CON) or containing DMSO (0.1%), 0.1 μM SQ1, 1 μM CITCO, or 100 μM PB. Culture medium was replaced once after 24 hours, and hepatocytes were harvested for the measurement of luciferase activity 48 hours after initial treatment. Each bar represents the mean ± S.E.M. of luciferase measurements (Gaussia/firefly) normalized to the untreated control group (CON). Shown are results combined from four independent hepatocyte preparations (n = 3 wells/treatment per hepatocyte preparation). *Significantly different compared with untreated (CON) controls (P < 0.05). †Significantly different compared with DMSO controls (P < 0.05).
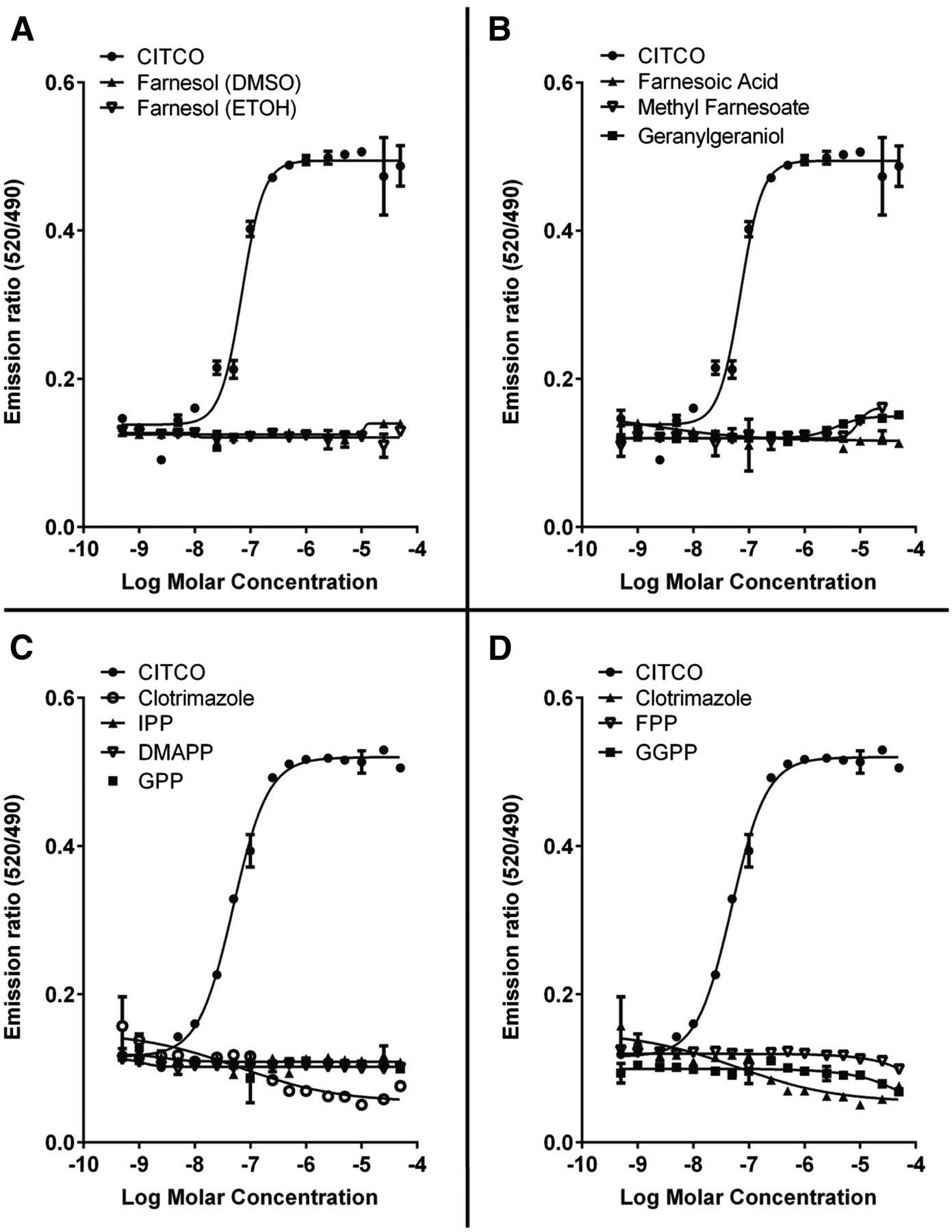
FOH and Nonsterol Isoprenoids Are Not Direct Ligand Agonists of hCAR1.
The failure of SQ1 treatment to induce a significant interaction between hCAR1 and SRC1, as was seen for the CAR ligand agonist CITCO, suggested that SQ1 treatment may not activate hCAR1 by causing accumulation of an hCAR ligand. To test whether FOH or several other isoprenoids could interact directly with the hCAR1 LBD, we used the LanthaScreen TR-FRET hCAR Coactivator Assay (Fig. 6). Whereas the positive control hCAR ligand agonist CITCO bound to hCAR1 with an average EC50 of 60.1 nM, and the hCAR1 inverse agonist clotrimazole bound with an IC50 of 60.2 nM, FOH showed no ability to bind to hCAR1 at concentrations up to 50 μM, whether added in DMSO or ETOH as the vehicle. Several other farnesoids (i.e., methyl farnesoate and farnesoic acid), isoprenoid pyrophosphates (i.e., dimethylallyl pyrophosphate, isopentenyl pyrophosphate, geranyl pyrophosphate, FPP, and geranylgeranyl pyrophosphate), and isoprenoid alcohols (geranylgeraniol) were also tested for their abilities to bind to hCAR1, and all showed little or no ability to bind (Fig. 6), although at the highest concentrations tested, geranylgeraniol and methyl farnesoate showed slight trends toward agonist-type binding, whereas FPP and geranylgeranyl pyrophosphate showed slight trends toward inverse agonist-type binding.

Evaluation of isoprenoid interactions with hCAR1-LBD in vitro. The LanthaScreen TR-FRET CAR Coactivator Assay was used to evaluate the ability of FOH and several other isoprenoids to interact with hCAR1. Stock solutions of FOH were prepared in either DMSO or ETOH; other compounds were prepared as described in Materials and Methods. CITCO was included as a positive control hCAR1 agonist, and clotrimazole was included as a positive control inverse agonist. Each point represents the mean ± S.D. of triplicate determinations, and binding curves were generated using Prism 6. The same binding curve for CITCO is shown in (A) and (B) [EC50 70.9 nM, 95% confidence interval (CI) 61.9–81.2 nM], and the same binding curves for CITCO and clotrimazole are shown in panels (C) and (D) (CITCO EC50 49.3 nM, 95% CI 45.5–53.3 nM; clotrimazole IC50 60.2 nM, 95% CI 12.2–299 nM). IPP, isopentenyl pyrophosphate; DMPP, dimethylallyl pyrophosphate; GPP, geranyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate.
Discussion
Nonsterol isoprenoids are intermediates produced in the cholesterol biosynthetic pathway that serve as substrates for prenylation reactions and are precursors for a number of important cellular products (Edwards and Ericsson, 1999). FOH and geranylgerianol, in particular, are biologically active molecules that can influence the expression of drug- and lipid-metabolizing enzymes through modulation of nuclear receptor signaling pathways (Kocarek and Mercer-Haines, 2002; Takahashi et al., 2002; Goto et al., 2011a). We previously reported that accumulation of isoprenoids in response to squalene synthase inhibition increased CYP2B1 expression in primary rat hepatocytes in a CAR-dependent manner (Kocarek et al., 1998; Kocarek and Mercer-Haines, 2002). In a continuation of these studies, we investigated whether mCAR or any of the major hCAR isoforms are activated by similar mechanisms. Using primary hepatocytes from wild-type, CAR-KO, and hCAR-TG mice, we found that squalene synthase inhibition significantly induced Cyp2b10 mRNA levels in mCAR- or hCAR-expressing hepatocytes, whereas this effect was attenuated in the absence of CAR. Consistent with previous findings (Kocarek and Reddy, 1996), pravastatin had no effect on Cyp2b10 expression, although both pravastatin and SQ1 induced Hmgcr mRNA to a similar extent. These findings indicate that the main effects of SQ1 on Cyp2b10 induction are mediated through CAR, and that both human and murine CAR are activated by endogenous isoprenoids.
The splice variants hCAR2 and hCAR3 are estimated to account for up to ∼10 and 50% of total hCAR transcripts in human liver (Jinno et al., 2004; Zhang et al., 2013), respectively, which was corroborated in the current study. The amino acid insertion into hCAR2 is predicted to alter CAR’s ligand binding pocket, thereby modifying the activation profile (DeKeyser et al., 2009). In comparison, ligand selectivity of hCAR3 is similar to that of the reference receptor (Omiecinski et al., 2011). In the current study, we evaluated the hCAR isoforms for the ability to activate CYP2B reporter activity and found that the effects of SQ1 were mediated primarily in an hCAR1-dependent manner. The normalized activation of hCAR1 by SQ1 was significant and only slightly less than that observed with either the ligand agonist CITCO or the indirect CAR activator PB. In comparison, CITCO did not strongly induce CYP2B reporter activity above background in the presence of either hCAR2 or hCAR3. Consistent with previous findings (Auerbach et al., 2003; Lau et al., 2011), however, CITCO was capable of activating all three receptors in the order hCAR1 > hCAR3 > hCAR2 as indicated by increased interaction with the hSRC1-RID. Both hCAR2 and hCAR3 are reported to have compromised ability to heterodimerize with RXR (Auerbach et al., 2005, 2007). This, together with our current findings, indicate weak transactivation potential on CYP2B response elements in the absence of exogenously added RXR (Arnold et al., 2004; Jinno et al., 2004; Auerbach et al., 2005), in the context of our mouse primary hepatocyte model.
Several nonsterol isoprenoids are known agonists for nuclear receptors, including thyroid hormone receptor (FPP), estrogen receptor (FPP), PPARα (FOH, geranylgeraniol), and PPARγ (FPP, FOH, geranylgeraniol) (Takahashi et al., 2002; Das et al., 2007; Goto et al., 2011b; Nagai et al., 2011). In the current study, FOH also induced CYP2B6-PBREM/XREM reporter activity in the presence of either mCAR or hCAR1; however, SQ1 did not increase the interaction of hCAR1 with the hSRC1-RID, suggesting that SQ1’s mechanism of CAR activation differs from that of the ligand agonist CITCO. We additionally evaluated the ability of FOH and several other isoprenoids to bind the hCAR1-LBD in vitro as indicated by association with a peptide representing the RID of a different coactivator (PCG1α). However, none of the compounds functioned as ligand agonists of hCAR1, further supporting an indirect mechanism of activation. The release of CAR from its cytoplasmic complex by either direct or indirect activators is thought to involve receptor dephosphorylation (Mutoh et al., 2009; Yang and Wang, 2014). Recently, PB and several flavonoids were demonstrated to mediate CAR dephosphorylation by antagonizing epidermal growth factor receptor signaling, indicating this pathway may be a common target for indirect CAR activation in the liver (Mutoh et al., 2013; Carazo Fernandez et al., 2015).
The findings that both mCAR and hCAR1 are activated by isoprenoid accumulation indicate this signaling pathway is conserved across species, although the magnitude of induction in mouse hepatocytes was severalfold lower than that previously reported in rat hepatocytes (Kocarek et al., 1998; Kocarek and Mercer-Haines, 2002). The reason for this difference is not known, but some possibilities include species differences in basal levels of CAR as well as functional crosstalk with other transcription factors, such as PPAR (Guo et al., 2007; Cheng and Klaassen, 2008; Wada et al., 2009; Saito et al., 2010). For example, FOH, geranylgeraniol, and farnesoic acid are activators of PPARα (O’Brien et al., 1996; Takahashi et al., 2002), and FOH metabolism is at least partially responsible for the triglyceride-lowering properties of squalene synthase inhibitors (Hiyoshi et al., 2003). Saito et al. (2010) reported that several synthetic PPARα agonists increased CAR and CYP2B1/2 mRNA levels by 5- to 10-fold in primary rat hepatocytes, which was dependent on CAR protein synthesis. The PPARα agonist WY14643 also enhanced PB-induced CYP2B1/2 expression in vivo as well as in primary rat hepatocytes through a functional PPRE in the rat CAR promoter, indicating activators of PPAR potentiate CAR responses in rats (Wieneke et al., 2007).
In mice, however, synthetic PPAR agonists attenuated both basal (unpublished observations) and PB-induced Cyp2b10 expression in the liver, probably through competition for coactivators (Guo et al., 2007). Inverse associations of CAR and PPAR on target gene expression have also been demonstrated in studies using knockout mice (Maglich et al., 2009); therefore, the overall extent of CAR induction may depend on PPAR activation as well as the coactivators recruited. Another mechanism could be species-dependent differences in isoprenoid metabolism (Kocarek and Mercer-Haines, 2002). FOH is hypothesized to be more rapidly metabolized in mice than rats (Vaidya et al., 1998), which could influence the concentration of metabolite(s) and/or signaling pathways driving CAR activation.
Limited information is available regarding hCAR2- and hCAR3-mediated promoter activation. Although both isoforms are thought to be ligand-dependent, some studies also support indirect activation mechanisms. For example, Lau and Chang (2013) reported that 3-hydroxyflavone induced both hCAR2 (SV23)- and hCAR3 (SV24)-driven reporter activity in the presence of RXR in HepG2 cells by a mechanism that did not involve recruitment of coactivators or transactivation of the LBD. Likewise, the indirect hCAR1 activators phenytoin and PB induced hCAR3-dependent activity of a (NR1)2-containing reporter in transfected HepG2 cells (Faucette et al., 2007). On the basis of these findings, it is plausible that FPP-derived isoprenoids may still influence activity of other hCAR isoforms, although, at least in the primary mouse hepatocyte model, this does not strongly activate CYP2B transcription. Given the relatively high expression of the major hCAR isoforms in human liver, future studies focusing on differences in global gene regulation between isoforms would be useful to determine whether there are more isoform-selective target genes and/or additional elements necessary for stronger transactivation.
In conclusion, results from the current study extend previous findings and indicate that FPP-derived isoprenoids activate CAR-mediated CYP2B responses, further linking cholesterol biosynthesis to CAR regulation among different species. Additional findings from coactivator recruitment assays further suggest that the effect of isoprenoids on mediating CAR responses occurs in an indirect manner. Endogenous isoprenoid levels are known to be influenced by a number of pharmaceutical drugs, including squalene synthase inhibitors, N-bisphosphonates, as well as statins (Park et al., 2014). Therefore, our findings have implications for future studies aimed at evaluating the biologic effects of these compounds on CAR-mediated signaling pathways as well as understanding species differences on hepatocellular physiology.
Acknowledgments
The authors thank Mary Gargano for technical skills in isolating the mouse primary hepatocytes; and Drs. Curtis Omiecinski (Pennsylvania State University), Thomas Chang (University of British Columbia), and James Granneman (Wayne State University) for providing plasmids that were essential for this study.
Authorship Contributions
Participated in research design: Rondini, Kocarek.
Conducted experiments: Rondini, Duniec-Dmuchowski.
Performed data analysis: Rondini, Duniec-Dmuchowski, Kocarek.
Wrote or contributed to the writing of the manuscript: Rondini, Duniec-Dmuchowski, Kocarek.
Footnotes
- Received December 7, 2015.
- Accepted January 20, 2016.
Dr. Rondini was funded, in part, through a postdoctoral fellowship awarded through the Office of Vice President of Research (Wayne State University, Detroit, MI). This research was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01 HL050710] and the National Institute of Environmental Health Sciences [Center Grant P30 ES020957].
Abbreviations
- CAR
- constitutive androstane receptor (NR1I3)
- CAR-KO
- CAR-knockout
- CAR-WT
- CAR–wild-type
- CITCO
- 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime
- CYP2B
- cytochrome P450 2B
- DMSO
- dimethyl sulfoxide
- ETOH
- ethanol
- FOH
- farnesol
- FPP
- farnesyl pyrophosphate
- GLuc-N
- Gaussia princeps luciferase N-terminal
- GLuc-C
- Gaussia princeps luciferase C-terminal
- hCAR
- human constitutive androstane receptor (NR1I3)
- hCAR1
- human CAR reference sequence (NCBI Reference Sequence NM_005122)
- hCAR2
- human CAR, splice variant 2 (NCBI Reference Sequence NM_001077480, alternatively named SV3 or SV23)
- hCAR3
- human CAR, splice variant 3 (NCBI Reference Sequence XM_005245697, alternatively named SV2 or SV24)
- hCAR-TG
- hCAR transgenic
- HMGCR
- 3-hydroxy-3-methylglutaryl-coenzyme A reductase
- LBD
- ligand binding domain
- mCAR
- murine CAR
- PB
- phenobarbital
- PBRE
- phenobarbital-responsive element
- PBREM
- phenobarbital-responsive enhancer module
- PPARα
- peroxisome proliferator–activated receptor alpha (NR1C1)
- PPARγ
- peroxisome proliferator–activated receptor gamma (NR1C3)
- PPRE
- peroxisome proliferator–activated receptor response element
- PCR
- polymerase chain reaction
- qRT-PCR
- quantitative reverse transcription–polymerase chain reaction
- RID
- receptor interaction domain
- RXR
- retinoid X receptor (NR2B)
- SQ1
- squalestatin 1 (zaragozic acid A)
- SRC
- steroid receptor coactivator (NCOA)
- Srebp
- sterol regulatory element-binding proteins
- SV
- splice variant
- TCPOBOP
- 1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene
- TR-FRET
- time-resolved fluorescence resonance energy transfer
- XREM
- xenobiotic-responsive enhancer module
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}