


Abstract
Recently, we cloned a human organic cation transporter, hOCT1, which is expressed primarily in the liver. hOCT1 plays an important role in the cellular uptake and elimination of various xenobiotics including therapeutically important drugs. HIV protease inhibitors are a new class of therapeutic agents. The purpose of this study was to elucidate the interactions of HIV protease inhibitors with hOCT1 and to determine whether hOCT1 is involved in the elimination of these compounds. We studied the interactions of HIV protease inhibitors with hOCT1 in a transiently transfected human cell line, HeLa. Uptake studies were carried out 40 h post-transfection using the radiolabeled model organic cation, [14C]tetraethylammonium (TEA), under different experimental conditions. In cis-inhibition studies, all of the HIV protease inhibitors tested, i.e., indinavir (IC50 of 62 μM), nelfinavir (IC50 of 22 μM), ritonavir (IC50 of 5.2 μM), and saquinavir (IC50 of 8.3 μM) inhibited TEA uptake in HeLa cells expressing hOCT1. However, none of the HIV protease inhibitorstrans-stimulated [14C]TEA uptake, suggesting that they are poorly translocated by hOCT1. Nelfinavir, ritonavir, and saquinavir demonstrated an apparent “trans-inhibition” effect. No enhanced uptake of [14C]saquinavir was observed in hOCT1 DNA-transfected cells versus empty vector-transfected cells. These data suggest that HIV protease inhibitors are potent inhibitors, but poor substrates, of hOCT1. Some HIV protease inhibitors may potently inhibit the uptake and elimination of cationic drugs that are substrates for hOCT1, leading to potential drug-drug interactions. Other transporters, e.g., MDR1 and MRP1, in HIV-targeted cells may control the intracellular concentrations of HIV protease inhibitors.
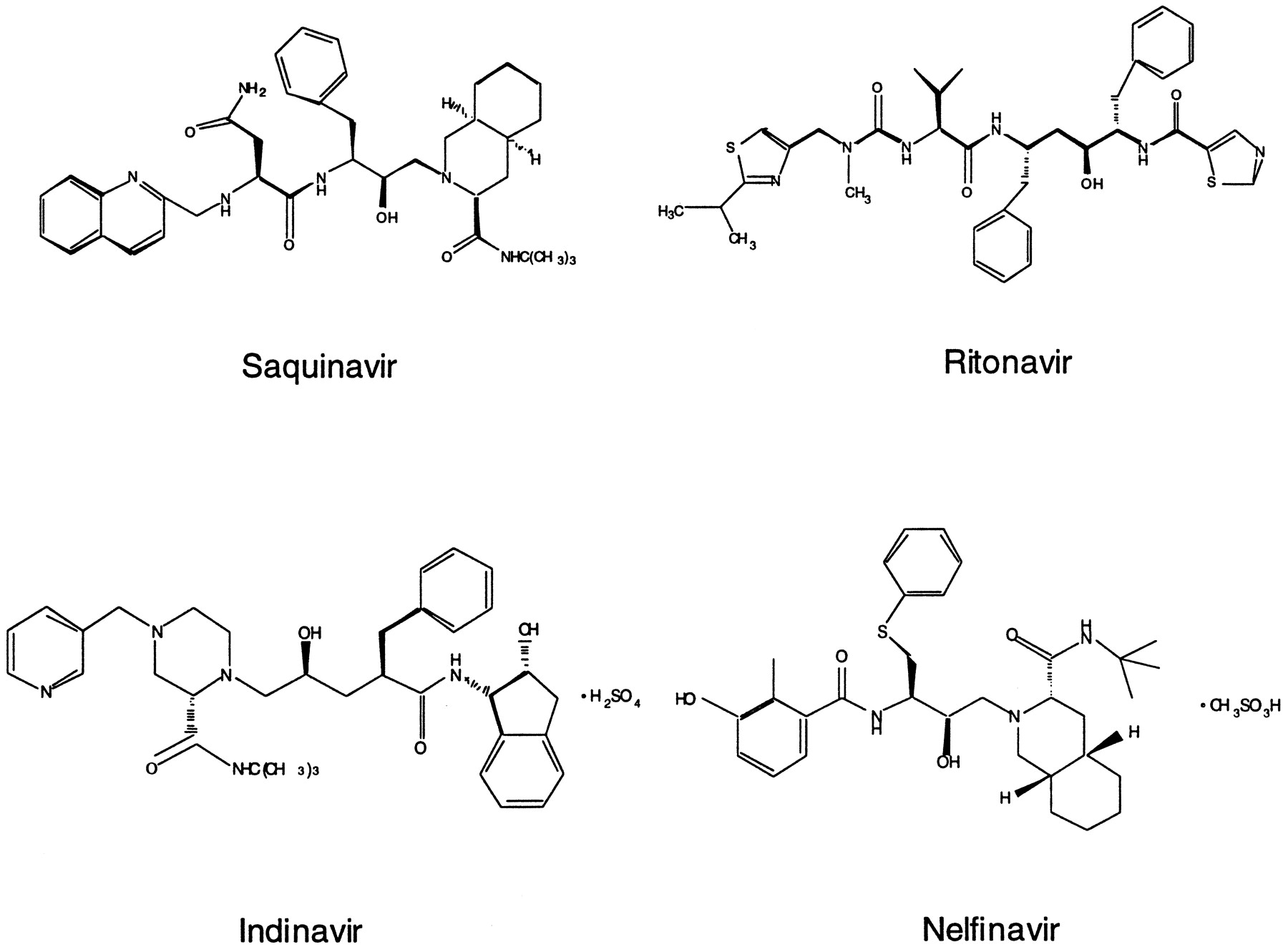
HIV protease is a virally encoded enzyme that is essential in the production of mature, infectious HIV virion particles. As soon as the HIV protease enzyme was recognized as a unique target for the treatment of AIDS, HIV protease inhibitors were developed and they represent a new class of therapeutically important drugs (Deeks and Volberding, 1997; McDonald and Kuritzkes, 1997; Pakyz and Israel, 1997; Kekuda et al., 1998). HIV protease inhibitors are peptidomimetics that competitively bind to the HIV protease active site and inhibit its proteolytic function, which leads to the production of immature, noninfectious HIV particles. HIV protease inhibitors are quite specific for the viral enzyme versus distantly related cellular enzymes, such as pepsin, renin, and cathepsin D, and are able to discriminate among them by several orders of magnitude (Deeks and Volberding, 1997; McDonald and Kuritzkes, 1997; Kakuda et al., 1998). Currently, four HIV protease inhibitors have been approved by the FDA for the treatment of AIDS: saquinavir, ritonavir, indinavir, and nelfinavir (Fig.1). These protease inhibitors have high potency in inhibiting HIV protease activity with IC95 (or EC95) values in the nanomolar ranges [saquinavir, 5–80 nM (EC90); ritonavir, 3.8–153 nM (EC50); indinavir, 25–100 nM; nelfinavir, 7–196 nM; drug package inserts].

Chemical structures of HIV protease inhibitors.
Generally, both metabolizing enzymes and xenobiotic transporters play a role in the elimination and detoxification of drugs. It is critical to understand which enzymes and transporters may interact with a drug to understand its pharmacokinetics and to predict potential drug-drug interactions. Saquinavir, ritonavir, and indinavir are primarily metabolized by cytochrome P-450 (CYP) 3A4, forming oxidative metabolites (Chiba et al., 1996, 1997; Kumar et al., 1996; Deeks and Volberding, 1997; Denissen et al., 1997; Fitzsimmons and Collins, 1997). Nelfinavir is metabolized by multiple P-450 isoforms including CYP3A [Wu et al., 1996; Nelfinavir (Viracept) package insert]. Recent data demonstrated that HIV protease inhibitors are substrates for the drug efflux pump, P-glycoprotein (P-gp), which may play an important detoxification role via countertransport of these agents (Kim et al., 1998a,b; Lee et al., 1998; Washington et al., 1998). It is not clear whether other xenobiotic transporters also play a role in the elimination of HIV protease inhibitors.
Organic cation transporters, often termed polyspecific organic cation transporters because of their broad substrate selectivity, are xenobiotic transporters that play a critical role in the disposition of various organic cations (Pritchard and Miller, 1993; Koepsell, 1998;Zhang et al., 1998a). Recently, we cloned a human organic cation transporter (hOCT), hOCT1, which is expressed primarily in the liver (Zhang et al., 1997). Functional studies in heterologous expression systems have demonstrated that various organic cations as well as other hydrophobic compounds interact with hOCT1 (Zhang et al., 1997, 1998b). Additional investigation of this uptake process is relevant in view of the major role of the liver in the distribution and elimination of cationic drugs and endogenous amines. Because HIV protease inhibitors carry positively charged amine moieties (Fig. 1), they may interact with organic cation transporters. In the present study, we investigated the interaction of these therapeutically important agents with hOCT1 in the transiently transfected HeLa cells. We carried out bothcis-inhibition [cis: test compounds on the same side of the membrane as the model substrate, [14C]tetraethylammonium (TEA)] andtrans-stimulation (trans: test compounds on the opposite side of the membrane as the model substrate, [14C]TEA) studies to determine both the interaction kinetics and substrate selectivity of hOCT1 for HIV protease inhibitors.
Materials and Methods
Materials.
HeLa cells and the media and buffers used to maintain the cells were purchased from the University of California San Francisco Cell Culture Facility. Original stocks of HeLa cells were obtained from American Type Culture Collection (ATCC, Rockville, MD). [14C]TEA (55 mCi/mmol) was purchased from American Radiolabeled Chemicals (St. Louis, MO). Indinavir sulfate and nelfinavir were purchased from the hospital pharmacy. Saquinavir mesylate was purchased from the hospital pharmacy and obtained in powdered form from Roche (Nutley, NJ) and [14C]saquinavir (3.30 mCi/mmol) was provided by Roche. Ritonavir was purchased from the hospital pharmacy and obtained in powdered form from Abbott (Abbott Park, IL). All other chemicals were obtained from Sigma (St. Louis, MO) and Fisher (Pittsburgh, PA) or as indicated.
DNA Isolation.
hOCT1 DNA subcloned into the mammalian expression vector pTargeT (Promega, Madison, WI) was used for transfection studies. Plasmid DNA was purified using the Qiagen Endo-free DNA isolation kit (Qiagen, Santa Clarita, CA). The DNA was stored in endotoxin-free TE buffer (10 mM Tris · Cl, 1 mM EDTA, pH 8.0) and its concentration was determined by UV spectroscopy. The yield from each isolation was approximately 400 to 900 μg with the DNA concentration ranging from 2.1 to 5.0 μg/μl in TE buffer.
HeLa Cell Culture and Transfection.
HeLa cells were maintained in the growth medium in 175-ml cell culture flasks (Nalge Nunc International, Naperville, IL) at 37°C in a humidified 5% CO2/95% air atmosphere as described previously (Zhang et al., 1998b). All studies were performed in cells between passages 2 and 20. Before transfection, cells were seeded at a density of 1.6 × 105 cells/well in 12-well tissue culture plates (Corning Costar Corp, Cambridge, MA). Following a modified protocol from Life Technologies, LipofectAMINE (2 mg/ml, Life Technologies) was used to deliver DNA to the cells (Zhang et al., 1998b). Briefly, for each well, 100 μl of Opti-MEM (Life Technologies) was mixed with 1 μg of DNA, and another 100 μl of media was mixed with 3.25 μl of lipid. The two solutions were then mixed and incubated for 30 min at room temperature. After incubation, 800 μl of Opti-MEM was added to the 200-μl mixture. The final volume of 1 ml of DNA/lipid complex mixture was applied to each well after rinsing the cells with 1 ml of the Opti-MEM. The cells were incubated for 18 h before the transfection mixture was replaced with standard complete growth medium.
Uptake Measurements.
Uptake studies were carried out 24 to 44 h post transfection as described previously (Zhang et al., 1998b). Briefly, the growth medium was gently removed, and each monolayer was rinsed with 1 ml of PBS buffer. To initiate uptake, 0.5 ml of PBS containing [14C]TEA (10 μM) was added to each well and incubated at room temperature for 20 min. In inhibition and IC50 studies, various concentrations of unlabeled compounds were included in the reaction mixture. The uptake was stopped by aspiration of the medium, and then each well of cells was immediately washed with ice-cold PBS buffer. The cells were solubilized with 1 ml of 0.5% Triton X-100, and 0.5 ml of sample was assayed by liquid scintillation counting (Beckman, Palo Alto, CA).
In trans-stimulation studies, each well of cells was preincubated with either 0.5 ml of PBS buffer (control) or 0.5 ml of PBS buffer plus the indicated amount of an unlabeled test compound at 37°C for 1 h. Cells were then rinsed with ice-cold PBS buffer before the uptake studies were performed as described above.
The protein concentration was determined by the Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA) with BSA as the standard as described previously (Zhang et al., 1998b).
Expression of hOCT1 in Xenopus laevis Oocytes and Transport Measurement.
Oocytes were harvested and maintained as described previously (Zhang et al., 1997). Fifty nanograms of hOCT1 cRNA was injected. Uptake studies were carried out 3 days postinjection using methods described in Zhang et al. (1997).
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR).
The first strand cDNA of MOLT-4 cells for PCR amplification was purchased from Clontech (Palo Alto, CA). The primers used in PCR were designed from the cDNA of hOCT1 (5′- and 3′-end primers in Zhang et al., 1997), MDR1 (sense and antisense primers were designed to amplify from positions 2419–2683 bp of the MDR1 cDNA, Genbank accession no.M14758) (Chen et al., 1986; Ueda et al., 1987) and MRP1 (sense and antisense primers were designed to amplify from 273–615 bp of the MRP1 cDNA, Genbank accession no. L05628) (Cole et al., 1992). PCR was performed in a thermal cycler (Perkin-Elmer, Foster City, CA) using the cycle as described previously (Zhang et al., 1997). The PCR products were electrophoresed through 1% agarose gel and stained with ethidium bromide.
Data Analysis.
In general, uptake values are expressed as mean ± S.E. or mean ± S.D. as indicated in the figure legends. A minimum of two wells were used to generate a data point in each experiment. All experiments were repeated at least once on a different day using a different cell passage. Statistical analysis was carried out by an unpaired Student's t test (Primer of Biostatistics software, Version 3, written by Stanton A. Glantz, McGraw-Hill Companies, 1991). Results were considered statistically different with a P < .05.
For IC50 studies, data were fit to the equationV = V0/[1 + (I/IC50)n] whereV is the uptake of [14C]TEA (20 min) in the presence of inhibitor, V0 is the uptake of [14C]TEA in the absence of inhibitor, I is the inhibitor concentration and n is the Hill coefficient. The Kaleidagraph fitting program (Abelbeck Software) was used to fit the data by nonlinear regression.
Results
Cis-Inhibition Studies.
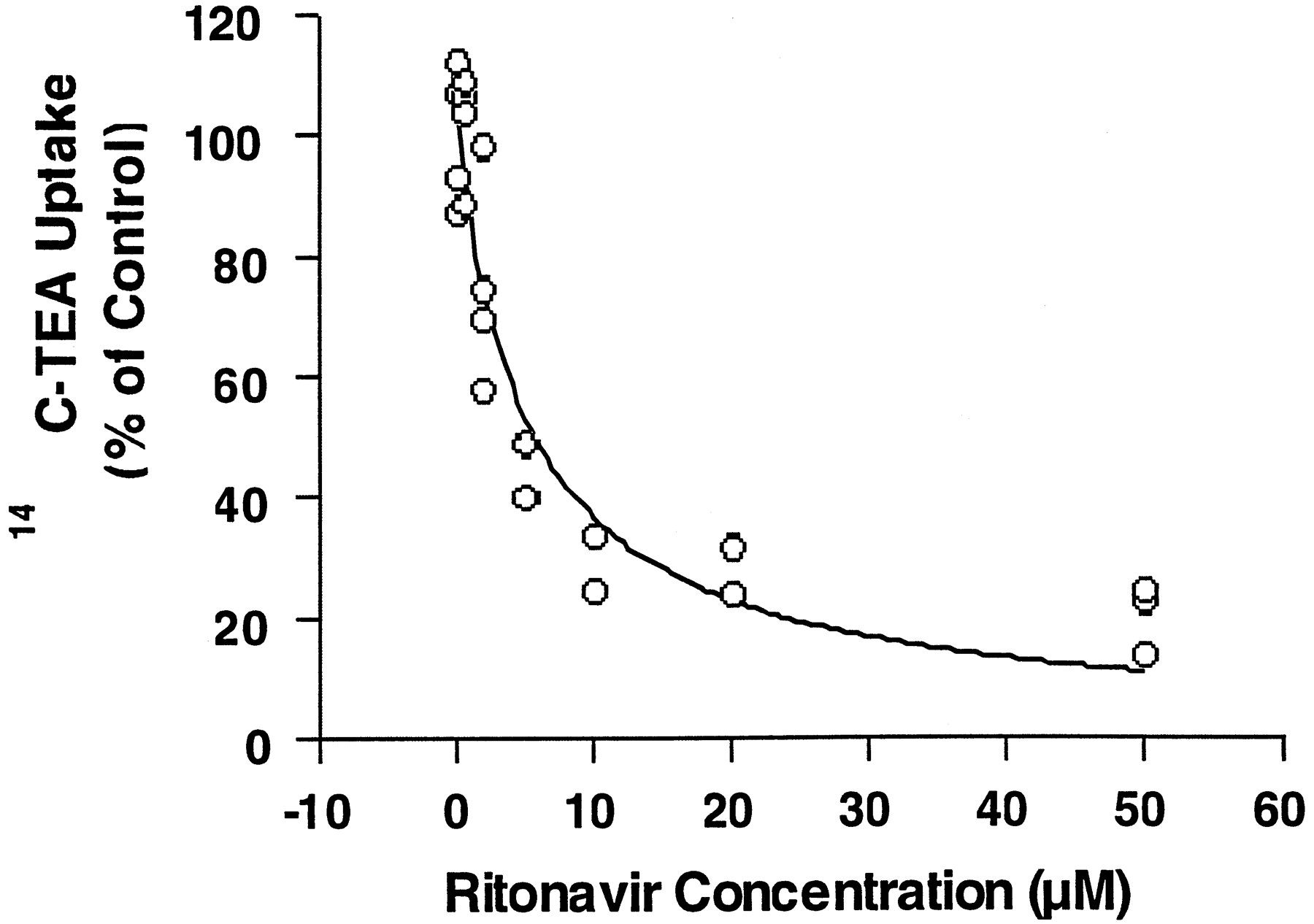
Uptake (accumulation) of [14C]TEA was measured at initial times when influx rate is much greater than efflux rate. Therefore, uptake rate is approximately the same as influx rate. The concentration dependence of the HIV protease inhibitors in inhibiting [14C]TEA uptake was determined. All HIV protease inhibitors significantly inhibited [14C]TEA uptake in HeLa cells expressing hOCT1. Data from multiple experiments were fit to the equation as described in the data analysis by nonlinear regression to obtain apparent IC50 values. Figure2 demonstrates a representative inhibition curve for ritonavir. IC50 values for these protease inhibitors ranged from 5.2 μM for ritonavir to 62 μM for indinavir; n values ranged from 0.59 for indinavir to 0.95 for ritonavir (Table 1).

Concentration-dependent inhibition of [14C]TEA uptake by ritonavir in HeLa cells transfected with pTargeT-hOCT1.
Initial rates of transport were determined in the presence of various concentrations of ritonavir. Data represent determinations from three experiments and were fitted with nonlinear regression as described inMaterials and Methods. The IC50 value of ritonavir obtained from the fit was 5.18 ± 1.21 μM.
IC50 values of HIV protease inhibitors inhibiting [14C]TEA uptake in hOCT1 DNA-transfected HeLa cells
trans-Stimulation Studies.
Inhibition does not imply that a compound is also translocated by a transporter. trans-Stimulation studies were used to determine whether the HIV protease inhibitors were substrates of hOCT1, i.e., translocated by hOCT1. In previous studies, we determined thattrans-stimulation is concentration-dependent, i.e., thetrans-stimulation effect increased with increasingtrans-TEA concentrations and it was saturable at high concentrations (concentrations ≥ 10 times theKm or Kivalues) (Zhang et al., 1999). Therefore, we assumed that at high concentrations, the maximum trans-stimulation effect for [14C]TEA uptake will be produced for any compound. After preincubating hOCT1 DNA-transfected HeLa cells with 2 mM TEA (“TEA” in Fig. 3) for 1 h at 37°C, [14C]TEA uptake was significantly enhanced (P < .05) in hOCT1 DNA-transfected cells preincubated with TEA in comparison with cells preincubated with buffer only (“Control” in Fig. 3). Preincubation of hOCT1 DNA-transfected cells with 200 μM indinavir did not result in a significant change in TEA uptake, whereas preincubation of cells with 200 μM nelfinavir, ritonavir, or saquinavir resulted in a significant decrease (apparent “trans-inhibition”) in [14C]TEA uptake (P < .05; Fig. 3). Similar results were obtained after incubation of the hOCT1 DNA-transfected cells with a lower concentration (50 μM) of unlabeled compounds (data not shown), suggesting that the apparent trans-inhibition was due to a decrease in the turnover rate of the transporter.

trans-Stimulation of [4C]TEA uptake in pTargeT-hOCT1-transfected HeLa cells.
The uptake (at 20 min) of [14C]TEA (10 μM) was measured after a 60-min preincubation (followed by washing) of the hOCT1 DNA-transfected cells (▪) and empty vector-transfected cells (▥) with PBS (Control) or PBS containing 2 mM TEA or 200 μM indinavir, nelfinavir, ritonavir, or saquinavir, respectively at 37°C. Data represent the mean ± S.D. of duplicate determinations obtained from one representative experiment. *P < .05.
Indinavir is the weakest inhibitor among the four HIV protease inhibitors. trans-Stimulation studies were carried out after preincubating the cells with 1 mM indinavir (i.e., >10 times itsKi value). Uptake studies were carried out at two different time points. As shown in Fig.4, no trans-stimulation effect was observed in cells preincubated with 1 mM indinavir.
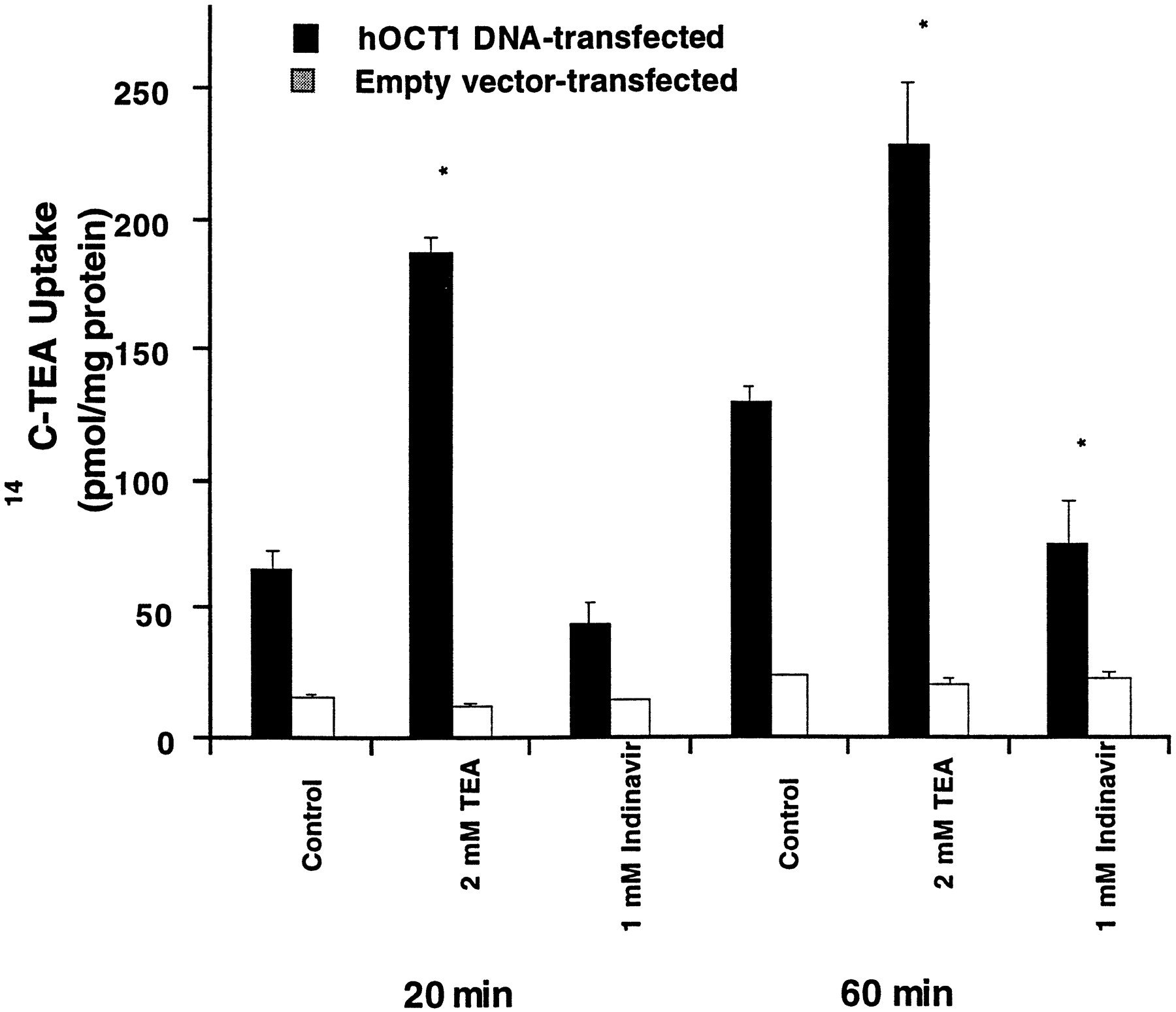
trans-Stimulation of [14C]TEA uptake in pTargeT-hOCT1 transfected HeLa cells.
The uptake (at 20 and 60 min) of [14C]TEA (10 μM) was measured after a 60-min preincubation (followed by washing) of the hOCT1 DNA-transfected cells (▪) and empty vector-transfected cells (□) with PBS (Control) or PBS containing 2 mM TEA or 1 mM indinavir, respectively at 37°C. Data represent the mean ± S.D. of duplicate determinations obtained from one representative experiment. *P < .05.
Permeant (Substrate) Studies.
A compound may or may not be a substrate if it does nottrans-stimulate or does trans-inhibit (Busch et al., 1998). Tracer influx experiments are required for a definite answer. Only radiolabeled saquinavir was available to us.14C-radiolabeled saquinavir uptake (26 μM) was directly measured in the hOCT1 DNA-transfected HeLa cells. As shown in Fig. 5, [14C]saquinavir uptake in hOCT1 DNA-transfected cells was not significantly different from that in empty vector-transfected cells, indicating that saquinavir was not translocated by hOCT1. As a positive control, [14C]TEA uptake (20 min) in the same experiment demonstrated a 3-fold enhanced uptake over that in empty vector-transfected cells at 20 min (data not shown).

Uptake of [14C]saquinavir (26 μM) in the pTargeT-hOCT1-transfected (●) and empty vector-transfected (○) HeLa cells.
Data represent mean ± S.D. of duplicate determinations from one representative experiment.
The endogenous saquinavir uptake in empty vector-transfected cells (496 pmol/mg protein/μM at 20 min) was high in comparison with endogenous TEA uptake (3.64 pmol/mg protein/μM at 20 min), thus an enhanced uptake of saquinavir might be masked by its high endogenous uptake. Accordingly, we determined the uptake of [14C]saquinavir (20 μM) in hOCT1 cRNA-injected oocytes, which have a low endogenous saquinavir uptake. We found that [14C]saquinavir uptake was not enhanced in hOCT1 cRNA-injected oocytes in comparison with water-injected oocytes (7.14 ± 1.13 versus 8.20 ± 2.34 pmol/oocyte/90 min, mean ± S.D.) 3 days postinjection when functional hOCT1 protein was expressed as assessed by [14C]TEA uptake.
Expression of hOCT1, MDR1, and MRP1 in MOLT-4 Cells.
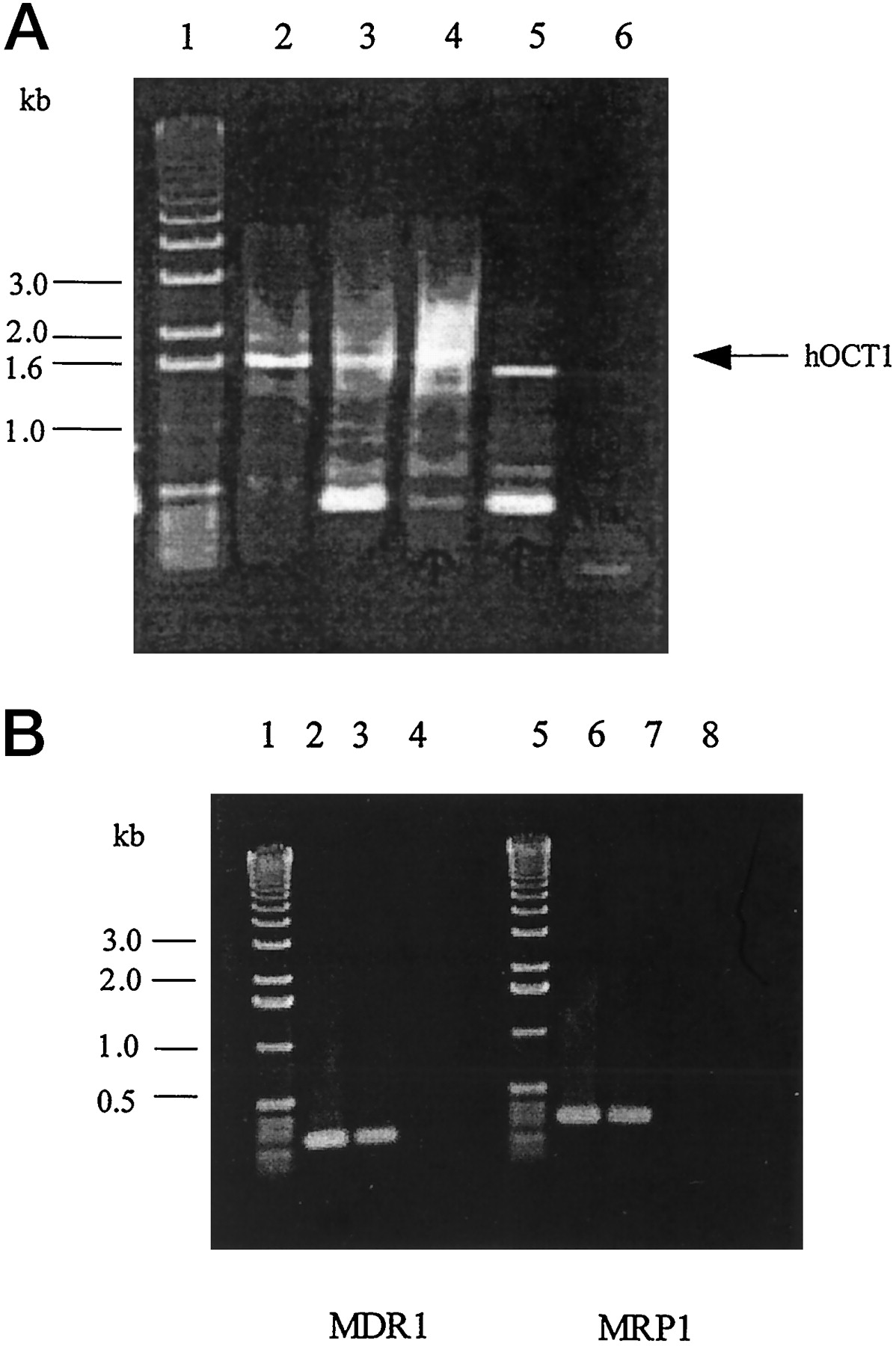
To explore the possibility that hOCT1 and other xenobiotic transporters may be important in the disposition of protease inhibitors in their target cells, the expression of the mRNA transcripts of the human hOCT1, MDR1, and MRP1 in MOLT-4 cells was determined. As shown in Fig.6, A and B, the mRNA transcripts of these transporters are expressed in MOLT-4 cells. However, RT-PCR using primers designed from hOCT1 cDNA resulted in amplifying an apparent splice variant of hOCT1. Lower bands in the gel may result from nonspecific amplification of DNA. Additional PCR using primers designed to amply the first half (5′-end) of hOCT1 and second half (3′-end) of hOCT1 DNA demonstrated that the deleted sequence was at the 3′-end of the cDNA (data not shown).

RT-PCR analysis of hOCT1, MDR1, and MRP1 mRNA transcript expression.
A, RT-PCR analysis of hOCT1 mRNA transcript expression in various tissues and cells. mRNA from human liver (lane 2), Caco-2 cells (lane 3), human small intestine (lane 4), or MOLT-4 cells (lane 5) was subjected to RT-PCR using primers designed to obtain a full-length DNA of the hOCT1 open reading frame (∼1.6 kb) as described inMaterials and Methods. Water (lane 6) was used as control in PCR reactions. The PCR products were analyzed by 1% agarose gel and stained with ethidium bromide. A 1-kb DNA ladder (Life Technologies) was used to determine the band size (lane 1). B, RT-PCR analysis of MDR1 and MRP1 mRNA transcript expression in MOLT-4 cells. Plasmid DNA containing MDR1 cDNA (lane 2, as positive control) or MRP1 cDNA (lane 6, as positive control) or mRNA from MOLT-4 cells (lanes 3 and 7) was subjected to RT-PCR as described in Materials and Methods. Water (lanes 4 and 8) was used as negative control in PCR reactions. The PCR products were analyzed by 1% agarose gel and stained with ethidium bromide. A 1-kb DNA ladder (Life Technologies) was used to determine the band size (lanes 1 and 5).
Discussion
HIV protease inhibitors are a new class of therapeutic agents for the treatment of AIDS. Although these drugs represent a clear advance in the management of HIV disease, drug resistance, side effects, and drug interactions are common. Both metabolizing enzymes and xenobiotic transporters play an important role in the elimination of these compounds and drug interactions can occur at both the metabolism and transport level. Furthermore, increasing evidence suggests that there is considerable overlap in the substrate selectivities of CYP3A4 and P-gp, indicating an increased potential for numerous drug interactions (Wacher et al., 1995, 1998; Benet et al., 1996; Schuetz et al., 1996a,b; Schinkel et al., 1997; van Asperen et al., 1997; Wacher et al., 1995, 1998; Washington et al., 1998; Zhang et al., 1998c). Previous studies have demonstrated that HIV protease inhibitors are inhibitors/substrates for CYP3A4 and P-gp (Chiba et al., 1996, 1997;Kumar et al., 1996; Deeks and Volberding, 1997; Fitzsimmons and Collins, 1997; Kim et al., 1998a,b; Lee et al., 1998; Lillibridge et al., 1998). Coadministration of protease inhibitors and drugs primarily metabolized by CYP3A4 and/or substrates for P-gp will result in altered plasma levels of other drugs, leading to adverse effects. For example, coadministration of rifampin, a known inducer of CYP3A4, with saquinavir decreased the steady-state AUC and maximum plasma concentration (Cmax) of saquinavir by approximately 80% [Saquinavir (Invirase) package insert].
To understand and predict pharmacokinetics and drug interactions, it is essential to determine the interaction of these agents with xenobiotic transporters. In the current study, interactions of HIV protease inhibitors with hOCT1 were investigated in hOCT1 DNA-transfected HeLa cells. We found that all four currently used HIV protease inhibitors are inhibitors of hOCT1 with IC50 values ranging from 5.18 μM for saquinavir to 61.7 μM for indinavir (Table 1). Human pharmacokinetics studies determined that steady-stateCmax of saquinavir, ritonavir, indinavir, and nelfinavir after oral dose are around 13, 6, 16, and 2 μM, respectively (Table 2). IC50 values determined in our studies are comparable to therapeutically relevant plasma concentrations (within 5-fold of steady-state Cmax values). These data suggest that HIV protease inhibitors, especially saquinavir and ritonavir, will potently interact with organic cation transport mediated by hOCT1.
Steady-state Cmax values of HIV protease inhibitors in comparison to their IC50 values inhibiting hOCT1
We further evaluated whether HIV protease inhibitors are transported by hOCT1. An indirect approach, trans-stimulation study (Holohan and Ross, 1980; Dantzler et al., 1991; Zhang et al., 1998b,1999), was used. If the test compound shares the same transporter as the model substrate (radiolabeled probe), the presence of the test compound on the opposite side (trans) of the membrane results in an enhanced flux of the radiolabeled probe. One possible mechanism is that the test compound inhibits the efflux of the model substrate resulting in an enhanced uptake. However, the absence oftrans-stimulation effect does not exclude the possibility that the test compound is a substrate (Busch et al., 1998). Tracer flux will provide a definite answer. Our trans-stimulation studies and permeant studies with [14C]saquinavir indicated that these compounds are poor substrates of hOCT1 (Figs. 3 and 5). Because HIV protease inhibitors are bulky molecules with molecular weights greater than 600, these data are consistent with our early findings that hydrophobic and bulky molecules are potent inhibitors but poor substrates of hOCT1 (Zhang et al., 1999).
Because HIV protease inhibitors are poorly translocated by hOCT1, it is likely that hOCT1 does not play a role in the elimination of these agents. However, these protease inhibitors (especially saquinavir and ritonavir) will potently inhibit the transport of cationic drugs, which are substrates of hOCT1 and lead to potential drug-drug interactions. For example, HIV protease inhibitors may inhibit the uptake and elimination of cationic drugs by the hepatocyte. Inhibition of uptake will lead to a poorer access of these drugs to metabolizing enzymes that are located inside the cells. Notably, the AUC andCmax of the antihistamine drug, terfenadine, were increased 358 and 253%, respectively, when coadministered with saquinavir (Fortovase) (package insert).
It is of considerable interest to determine the expression of xenobiotic transporters that may control the intracellular concentrations of a number of therapeutic agents including HIV protease inhibitors in HIV-target cells. CD4, the target for HIV virion infection, is expressed in MOLT-4 cells, a human lymphoblast leukemia cell line derived from the peripheral blood. Accordingly, we studied the expression of the xenobiotic transporters, hOCT1, MDR1, and MRP1, in MOLT-4 cells to determine their possible roles in HIV target cells. By RT-PCR analysis, we found that the multidrug resistant proteins, MDR1 (P-gp) and MRP1, are expressed in MOLT-4 cells (Fig. 6B). In addition, a putative spliced variant of hOCT1 is also expressed in this cell line (Fig. 6A). The expression of MDR1 in the MOLT-4 cells is consistent with previous studies that demonstrate that P-gp is expressed in CD4+, CD8+, CD56+, and CD19+ subsets of human peripheral blood lymphocytes (Chaudhary et al., 1992; Gupta et al., 1992; Lucia et al., 1995) and in H9 T cells and U937 monocyte cells (Gollapudi and Gupta, 1990). Azidothymidine and some nucleoside analogs are substrates for P-gp and the expression of this xenobiotic transporter has been associated with the resistance of HIV-infected T cells to these agents (Antonelli et al., 1992). With the recent observation that protease inhibitors are substrates for P-gp (Kim et al., 1998a,b; Lee et al., 1998), it seems plausible that this transporter may play a similar role in the resistance of CD4+ target cells to these agents by limiting their cellular access. To our knowledge, this is the first demonstration of MRP1 expression in T cells. Additional studies are needed to understand the functional role of MRP1 and the putative hOCT1 splice variants in protease inhibitor target cells.
Footnotes
-
Send reprint requests to: Kathleen M. Giacomini, Ph.D., Department of Biopharmaceutical Sciences, University of California San Francisco, 513 Parnassus, S-926, San Francisco, CA 94143-0446. E-mail:kmg{at}itsa.ucsf.edu
-
↵1 Present address: Metabolism and Pharmacokinetics, Bristol-Myers Squibb Company, Wallingford, Connecticut.
-
↵2 Present address: Department of Drug Metabolism, Genentech, Inc., South San Francisco, California.
-
This study was supported by National Institutes of Health Grants GM-57656 and GM-36780 (K.M.G.) and HL-53994 (D.L.K.). Lei Zhang was supported in part by the University of California San Francisco Chancellor's Graduate Research Fellowship.
- Abbreviations used are::
- TEA
- tetraethylammonium
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- Cmax
- maximum plasma concentration
- CYP
- cytochrome P-450
- hOCT1
- human organic cation transporter 1
- P-gp
- P-glycoprotein
- Received May 7, 1999.
- Accepted November 15, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}