


Abstract
To quantitatively understand the events in the human liver, we modeled a hepatic disposition of bosentan and its three known metabolites (Ro 48-5033, Ro 47-8634, and Ro 64-1056) in sandwich-cultured human hepatocytes based on the known metabolic pathway. In addition, the hepatotoxicity of Ro 47-8634 and Ro 64-1056 was investigated because bosentan is well known as a hepatotoxic drug. A model illustrating the hepatic disposition of bosentan and its three metabolites suggested the presence of a novel metabolic pathway(s) from the three metabolites. By performing in vitro metabolism studies on human liver microsomes, a novel metabolite (M4) was identified in Ro 47-8634 metabolism, and its structure was determined. Moreover, by incorporating the metabolic pathway of Ro 47-8634 to M4 into the model, the hepatic disposition of bosentan and its three metabolites was successfully estimated. In hepatocyte toxicity studies, the cell viability of human hepatocytes decreased after exposure to Ro 47-8634, and the observed hepatotoxicity was diminished by pretreatment with tienilic acid (CYP2C9-specific inactivator). Pretreatment with 1-aminobenzotriazole (broad cytochrome P450 inactivator) also tended to maintain the cell viability. Furthermore, Ro 64-1056 showed hepatotoxicity in a concentration-dependent manner. These results suggest that Ro 64-1056 is directly involved in bosentan-induced liver injury partly because CYP2C9 specifically mediates hydroxylation of the t-butyl group of Ro 47-8634. Our findings demonstrate the usefulness of a quantitative modeling of hepatic disposition of drugs and metabolites in sandwich-cultured hepatocytes. In addition, the newly identified metabolic pathway may be an alternative route that can avoid Ro 64-1056–induced liver injury.
Introduction
Sandwich-cultured hepatocytes (SCHs) have recently become a valuable in vitro tool for investigating the biliary excretion of endogenous and exogenous compounds, because SCHs form intact canalicular networks and have a polarized excretory function (Swift et al., 2010; Brouwer et al., 2013). In addition, SCHs retain other in vivo–like properties, including constitutively expressed enzyme activities and the entire array of transport proteins involved in basolateral uptake and efflux (Brouwer et al., 2013). Because of these features, the SCH system provides the overall hepatic disposition of a parent drug and its metabolites formed in hepatocytes by exploiting constitutively expressed enzyme activities (Matsunaga et al., 2013). Furthermore, model-based concepts have recently become widespread in various research fields, including drug discovery and development. Several model-based analyses have been undertaken for the hepatic disposition of drugs and their metabolites in human SCHs to predict intracellular concentration changes by alterations in hepatobiliary transport, to investigate rate-limiting transport processes, and to evaluate drug-drug interaction at clinically relevant concentrations (Lee et al., 2010; Matsunaga et al., 2014, 2015).
Bosentan is an oral nonselective endothelin receptor antagonist used for the treatment of pulmonary arterial hypertension (PAH) (Seferian and Simonneau, 2013). As shown in Fig. 1, bosentan is metabolized by CYP3A4 and CYP2C9 in the human liver to three metabolites: Ro 48-5033 (hydroxylation at the t-butyl group of bosentan), Ro 47-8634 (O-demethylation of the phenolic methyl ether of bosentan), and Ro 64-1056 (combination of hydroxylation and O-demethylation of bosentan) (van Giersbergen et al., 2002; Markova et al., 2013). Hepatic metabolism followed by biliary excretion of these metabolites is a major elimination pathway of bosentan in humans (Weber et al., 1999), meaning that the hepatic disposition of bosentan and its metabolites determines their fates in the body.
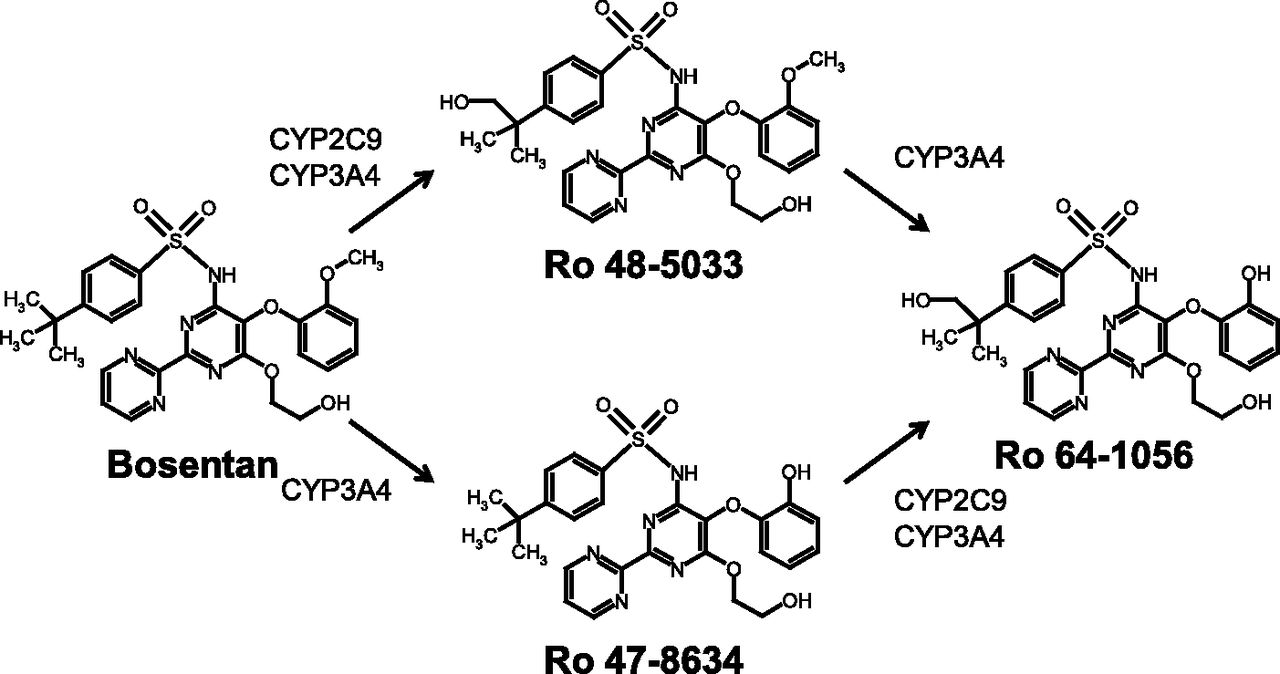
Known metabolic pathway of bosentan and its three metabolites in the human liver as proposed by van Giersbergen et al. (2002) and Markova et al. (2013).
Recent long-term safety data on bosentan from the Endthelin Antagonist tRial in miLdlY symptomatic PAH patients (EARLY) study showed that 16.8% of patients receiving bosentan experienced elevations of alanine and/or aspartate aminotransferases to more than 3 times the upper limit of normal over the 5-year duration of the study, and that most of the increases in alanine and/or aspartate amino transferases associated with bosentan occurred during the first 6 months of treatment (Simonneau et al., 2014). Since the hepatotoxic effects are reversible and dose-dependent (Fattinger et al., 2001; Rubin et al., 2002; Treiber et al., 2014), treatment discontinuation, dose reduction, or continued treatment coupled with monthly blood tests to monitor liver enzymes should be used in cases of liver enzyme abnormalities (Simonneau et al., 2014). Although the mechanisms underlying bosentan-induced liver injury are not yet fully understood, cholestatic liver injury is considered to be one of the hepatotoxic mechanisms of bosentan, and inhibition of hepatocanalicular bile salt export pump (BSEP) is at least partly involved (Fattinger et al., 2001; Dawson et al., 2012; Treiber et al., 2014). Bosentan potently inhibited both human BSEP– and rat Bsep–mediated taurocholate transport with similar IC50 values (Dawson et al., 2012). In addition, the Ki values for rat Bsep were similar between Ro 47-8634 (O-demethylated form of bosentan) and bosentan (Fattinger et al., 2001). However, a recent mechanistic model linked with BSEP inhibition–mediated hepatotoxicity underpredicted the liver injury induced by bosentan (Woodhead et al., 2014). The authors mentioned several possibilities for the discrepancy, including no assumption for inhibition of basolateral efflux transport of bile acid by bosentan and a mechanistic effect of bile acid and/or bosentan on mitochondrial activity. Similarly, this discrepancy may also be applied to bosentan’s metabolites, because Ro 47-8634 showed rat Bsep inhibition with a Ki value similar to that of bosentan. Therefore, to consider bosentan-induced liver injury, it may be necessary to incorporate the hepatotoxic potential of not only bosentan but also its metabolites into the mechanistic model. To date, bosentan itself showed dose-dependent cytotoxicity for human hepatocytes. In addition, among the metabolites of bosentan, the cytotoxicity of Ro 48-5033 (t-butyl hydroxylated form of bosentan) was negligible (Markova et al., 2013), whereas those of the other two metabolites (Ro 47-8634 and Ro 64-1056) remain unclear.
In the present study, we constructed a hepatic disposition model of bosentan and its three metabolites to evaluate their kinetics in three compartments, including intracellular space, bile canalicular lumen, and extracellular medium, using human SCHs. In addition, the cytotoxicity of two bosentan metabolites, Ro 47-8634 and Ro 64-1056 (not bosentan itself), was examined toward human hepatocytes.
Materials and Methods
Materials.
Bosentan (4-tert-butyl-N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)[2,2′-bipyrimidin]-4-yl]benzenesulfonamide), bosentan-d4 (4-tert-butyl-N-[6-(2-hydroxyethoxy-d4)-5-(2-methoxyphenoxy)[2,2′-bipyrimidin]-4-yl]benzenesulfonamide), Ro 48-5033 (4-(2-hydroxy-1,1-dimethylethyl)-N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)[2,2′-bipyrimidin]-4-yl]-benzenesulfonamide), Ro 48-5033-d4 (4-(2-hydroxy-1,1-dimethylethyl)-N-[6-(2-hydroxyethoxy-d4)-5-(2-methoxyphenoxy)[2,2′-bipyrimidin]-4-yl]-benzenesulfonamide), Ro 47-8634 (4-tert-butyl-N-[6-(2-hydroxyethoxy)-5-(2-hydroxyphenoxy)[2,2′-bipyrimidin]-4-yl]benzenesulfonamide), and Ro 64-1056 (4-(2-hydroxy-1,1-dimethylethyl)-N-[6-(2-hydroxyethoxy)-5-(2-hydroxyphenoxy)[2,2′-bipyrimidin]-4-yl]benzenesulfonamide) were purchased from Toronto Research Chemicals Inc. (North York, Canada). Tienilic acid (TA) was purchased from Corning Life Sciences (Tewkesbury, MA). Erythromycin (ETM) and 1-aminobenzotriazole (1-ABT) were purchased from Sigma-Aldrich (St. Louis, MO). Cryopreserved human hepatocytes (lots IZT, DQB, and GMX) were purchased from BioreclamationIVT (Baltimore, MD). Cryopreserved human hepatocytes (lots Hu1437 and Hu8110), human liver microsomes (lot PL050B, pool of 50 donors, mixed gender), cryopreserved hepatocyte recovery medium, cryopreserved hepatocyte plating medium, hepatocyte maintenance supplement packs, Geltrex, phenol red–free Williams’ medium E (WME), and Hanks’ balanced salt solution were purchased from Life Technologies Corporation (Carlsbad, CA). All other chemicals and reagents were of special or high-performance liquid chromatography grade. The characteristics of each hepatocyte lot are shown in Table 1.
Characteristics of cryopreserved human hepatocytes
Hepatic Disposition of Bosentan and Its Metabolites in Human SCHs.
Cryopreserved human hepatocytes were thawed, plated, and cultured as described in our previous study (Matsunaga et al., 2014), and human SCHs on day 4 were used. Modulation of Ca2+ in the incubation buffer was applied to maintain or disrupt the canalicular networks between cells sealed by tight junctions. Using this approach, the masses of compounds in the bile canalicular lumen can be measured with Ca2+-containing and Ca2+-free buffers (B-CLEAR technology; Qualyst Transporter Solutions, Durham, NC) (Brouwer et al., 2013). Metabolism and hepatobiliary transport studies of human SCHs were performed as described in our previous report (Matsunaga et al., 2014). In brief, human SCHs were incubated with Ca2+-containing or Ca2+-free buffer containing bosentan at an initial concentration of 1 µM at 37°C for 3, 5, 10, 15, 20, 30, or 45 minutes. After the incubation, the entire volume of the buffer was collected, and the cells were destroyed by adding ethanol.
Metabolism in Human Liver Microsomes.
Ro 48-5033 (0.5 µM), Ro 47-8634 (0.5 µM), or Ro 64-1056 (0.5 µM) was incubated with 0.5 mg/ml human liver microsomes at 37°C in a buffer (5 mM MgCl2, 100 mM potassium phosphate, pH 7.4) containing 1 mM NADPH for 0, 5, 10, 20, or 30 minutes. Ro 64-1056 (0.5 µM) was also incubated with 0.5 mg/ml human liver microsomes, which had been pretreated with 25 µg/ml alamethicin for 10 minutes on ice, in a buffer (8 mM MgCl2, 50 mM Tris-HCl, pH 7.5) containing 2 mM uridine 5′-diphosphoglucuronic acid (UDPGA) for 30 minutes at 37°C. After the incubation, 200 µl of the reaction mixture was transferred to a tube containing 1 ml of methanol/acetonitrile mixture (1:1, v/v) to stop the metabolism, and internal standards dissolved in methanol were added. The mixture was ultrasonicated and centrifuged, and the entire volume of the supernatant was collected in a glass tube. One milliliter of methanol/acetonitrile mixture (1:1, v/v) was added to the residue, and ultrasonication and centrifugation were performed again. The entire volume of the supernatant was collected and combined with the previous supernatant, and the mixed supernatant was evaporated to dryness. The residue was reconstituted with 10 mM ammonium acetate (pH 3.5)/acetonitrile (7:3, v/v), and the reconstituted sample was injected into a liquid chromatography with tandem mass spectrometry (LC-MS/MS) system.
Hepatotoxicity Assay.
Thawed human hepatocytes suspended in cryopreserved hepatocyte plating medium were plated in collagen I–coated 96-well plates. The medium was discarded at 4 hours after plating, and the hepatocytes were preincubated with TA (10 µM; specific CYP2C9 inactivator), ETM (10 µM; specific CYP3A inactivator), 1-ABT [1 mM; broad cytochrome P450 (P450) inactivator], or dimethylsulfoxide (0.1%; vehicle control) in WME for 20 hours. Subsequently, the medium was replaced with fresh WME containing Ro 47-8634 (0–200 µM), Ro 64-1056 (0–200 µM), or Triton X-100 (1%; positive control), and the plated hepatocytes were incubated for 48 hours with replacement of the medium every 24 hours. At the end of the incubation, the cell viability was assessed by the WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium) assay (Dojindo Laboratories, Kumamoto, Japan), in accordance with the manufacturer’s protocols. WST-8 is a water-soluble tetrazolium salt and is reduced to a colored water-soluble formazan by a mitochondrial enzyme, dehydrogenase, in the cells.
Analytical Methods.
An Agilent 1100 high-performance liquid chromatography system equipped with a binary pump, column oven, and autosampler (Agilent Technologies, La Jolla, CA) connected to an API4000 triple quadrupole mass spectrometer equipped with a turbo ion spray interface (AB Sciex, Foster City, CA) was used for LC-MS/MS analysis. The detailed methods for quantification and confirmation of the fragmentations are presented in the Supplemental Material. Analyst 1.4.1 software (AB Sciex) was used to control the LC-MS system and perform the sample data analyses.
Mathematical Modeling of the Hepatic Disposition of Bosentan and Its Metabolites.
Population pharmacokinetic analyses were performed using a nonlinear mixed-effect model program, NONMEM version 7.1.2 (subroutine ADVAN 13; Globomax, Hannover, MD), to fit compartmental models to the hepatic disposition of bosentan and its metabolites in human SCHs based on Fig. 3 (model 1) and Fig. 8 (model 2). The differential equations are detailed in the Supplementary Material.
Statistical Analysis.
SAS 9.2 software (SAS Institute Inc., Cary, NC) was used. Paired t tests were performed for the effects of pretreatment in hepatocyte toxicity studies. Dunnett’s tests were also used to determine statistical significance between treatment groups. A value of P < 0.05 was prespecified as the criterion for significance in each analysis.
Results
Metabolism and Hepatobiliary Transport of Bosentan and Its Metabolites in Human SCHs.
The amounts of bosentan, Ro 48-5033, Ro-47-8634, and Ro 64-1056 in three compartments, including the buffer, cells, and bile canalicular lumen, were measured by maintaining or disrupting tight junctions using Ca2+-containing or Ca2+-free buffer, respectively. Figure 2 shows the mass-time profiles of bosentan, Ro 48-5033, Ro-47-8634, and Ro 64-1056 in these three compartments. The accumulation of bosentan in cells reached a plateau after 15 minutes of incubation (Fig. 2A), and the difference between Ca2+-containing and Ca2+-free buffers, which represents the amount in bile, was less than 6% of total amounts of bosentan in the three compartments. Therefore, the rate of biliary excretion of bosentan was considered negligible for hepatic disposition of bosentan itself in human SCHs, and the biliary excretion process of bosentan was not incorporated into our hepatic disposition model. Bosentan’s three metabolites (Ro 48-5033, Ro 47-8634, and Ro 64-1056) increased time dependently in all three compartments (Fig. 2, B–D). Therefore, the biliary excretion and basolateral efflux processes of these metabolites were incorporated into the hepatic disposition model. The total recovered amounts of bosentan and its three metabolites were 88.3–111.9% of the dose.

Mass-time profiles of bosentan, Ro 48-5033, Ro 47-8634, and Ro 64-1056 from in vitro metabolism and hepatobiliary transport studies. Bosentan (1 µM) was incubated with human SCHs for 3, 5, 10, 15, 20, 30, or 45 minutes in Ca2+-containing or Ca2+-free buffer. The amounts (pmol) of bosentan (A), Ro 48-5033 (B), Ro 47-8634 (C), and Ro 64-1056 (D) were measured in the buffer (open circle), cells (closed square), and bile canalicular lumen (open triangle). Data represent means ± S.D. from three independent experiments with one lot of human hepatocytes (IZT).
Establishment of a Hepatic Disposition Model: Possibility of an Unidentified Metabolic Pathway of Bosentan’s Metabolites in the Human Liver.
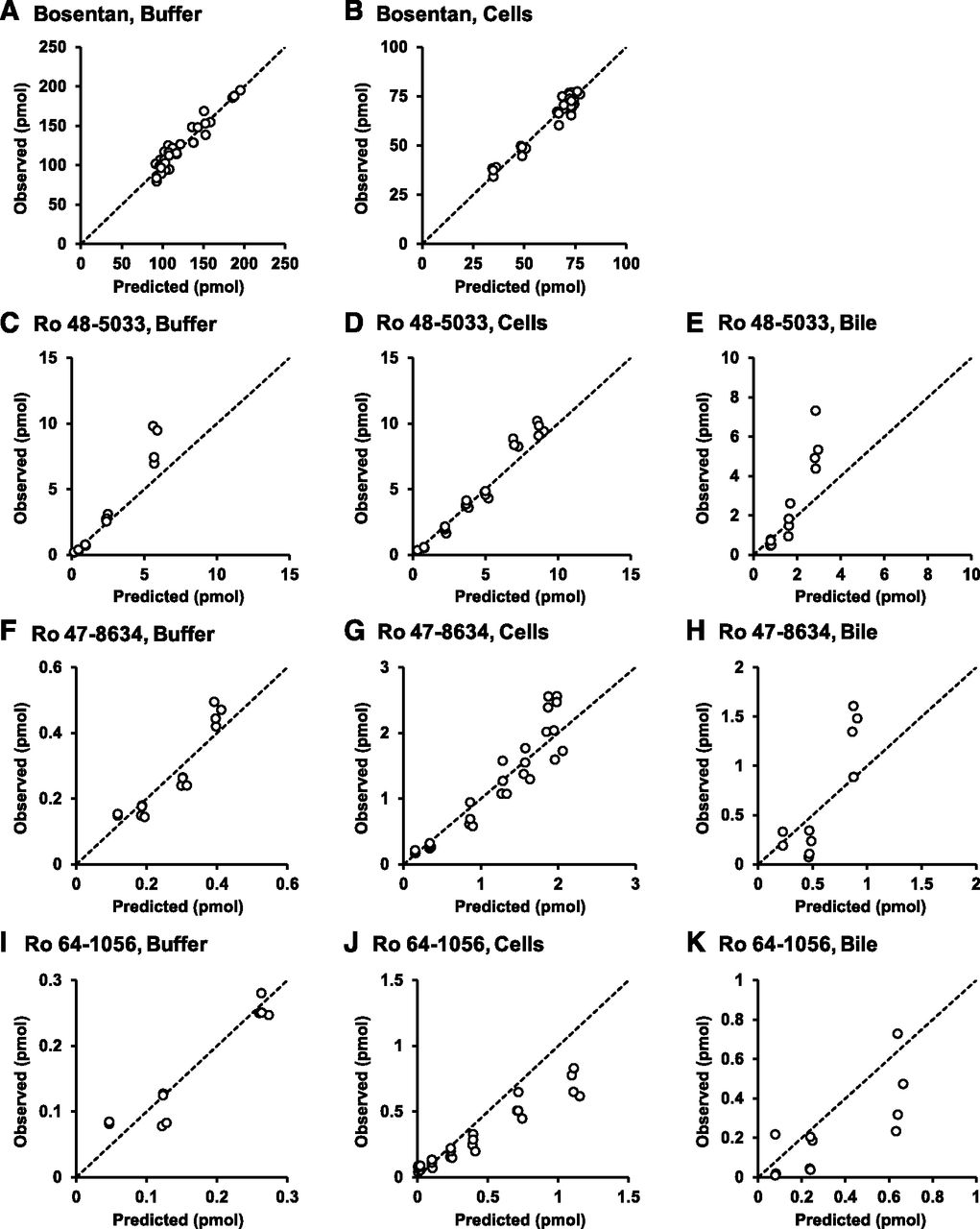
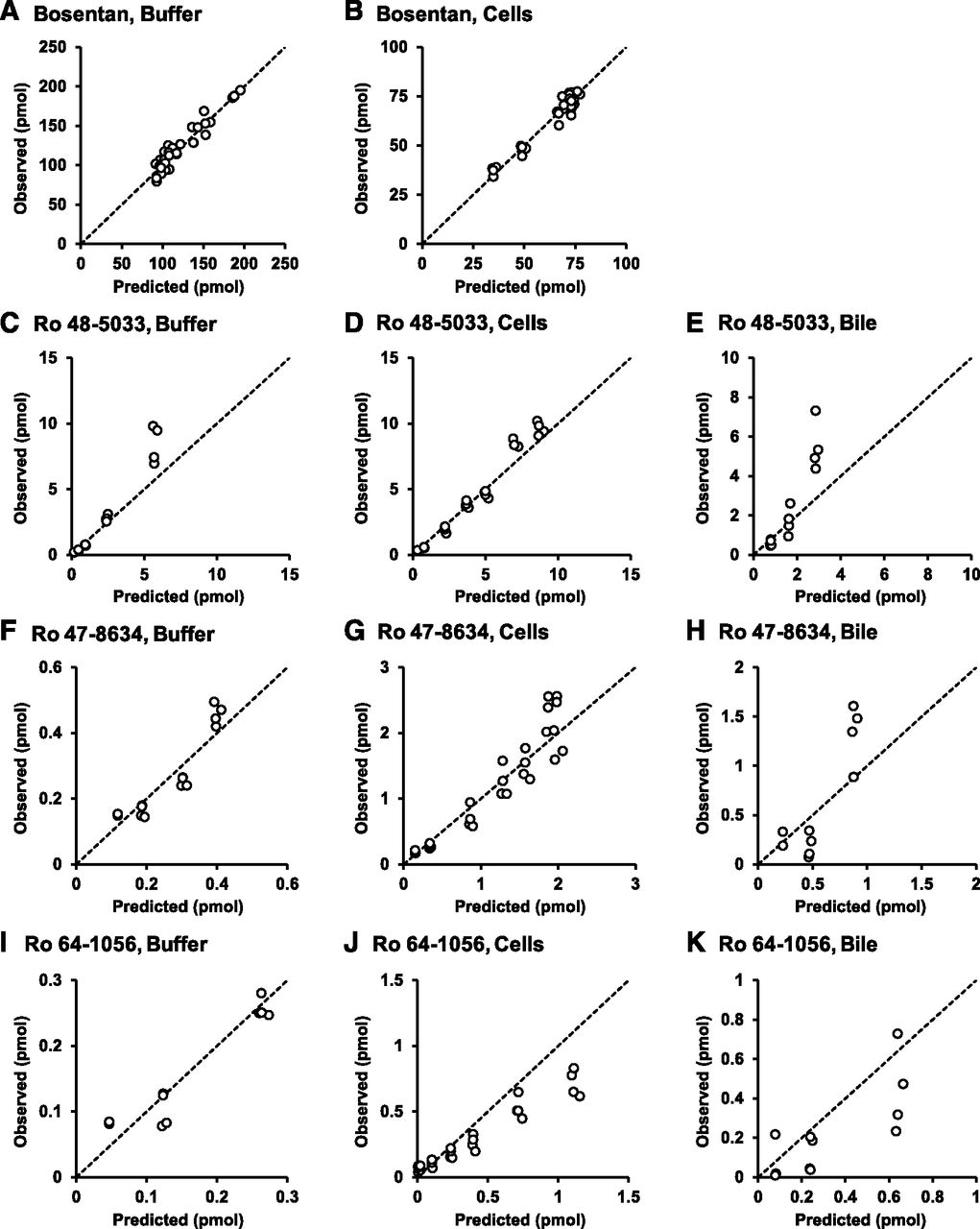
Based on the results obtained in Fig. 2, a hepatic disposition model 1 with the known metabolic pathways of bosentan (Fig. 3) was constructed. The first-order rate constants for basolateral uptake and efflux, intracellular metabolism, biliary excretion, and flow from the bile canalicular lumen to the buffer were estimated from the mass-time profiles of bosentan and its metabolites in the three compartments. Using the estimated parameters shown in Table 2, the correlations between the predicted and observed values were examined (Fig. 4). In the model, most of the observed values were uniformly distributed around the unity line. However, intracellular Ro 64-1056 was significantly overestimated (Fig. 4J). These results suggest that the overestimation of Ro 64-1056 in the cell compartment arose through the matching of the values in all other compartments. To explain such an overestimation, the following three hypotheses were considered: 1) Ro 64-1056 was further metabolized; 2) the metabolic rate from Ro 48-5033 to Ro 64-1056 was overestimated due to the presence of an unidentified metabolic pathway of Ro 48-5033, which was apparently involved in the metabolic rate from Ro 48-5033 to Ro 64-1056; and 3) the metabolic rate from Ro 47-8634 to Ro 64-1056 was overestimated for the same reason as in hypothesis 2.
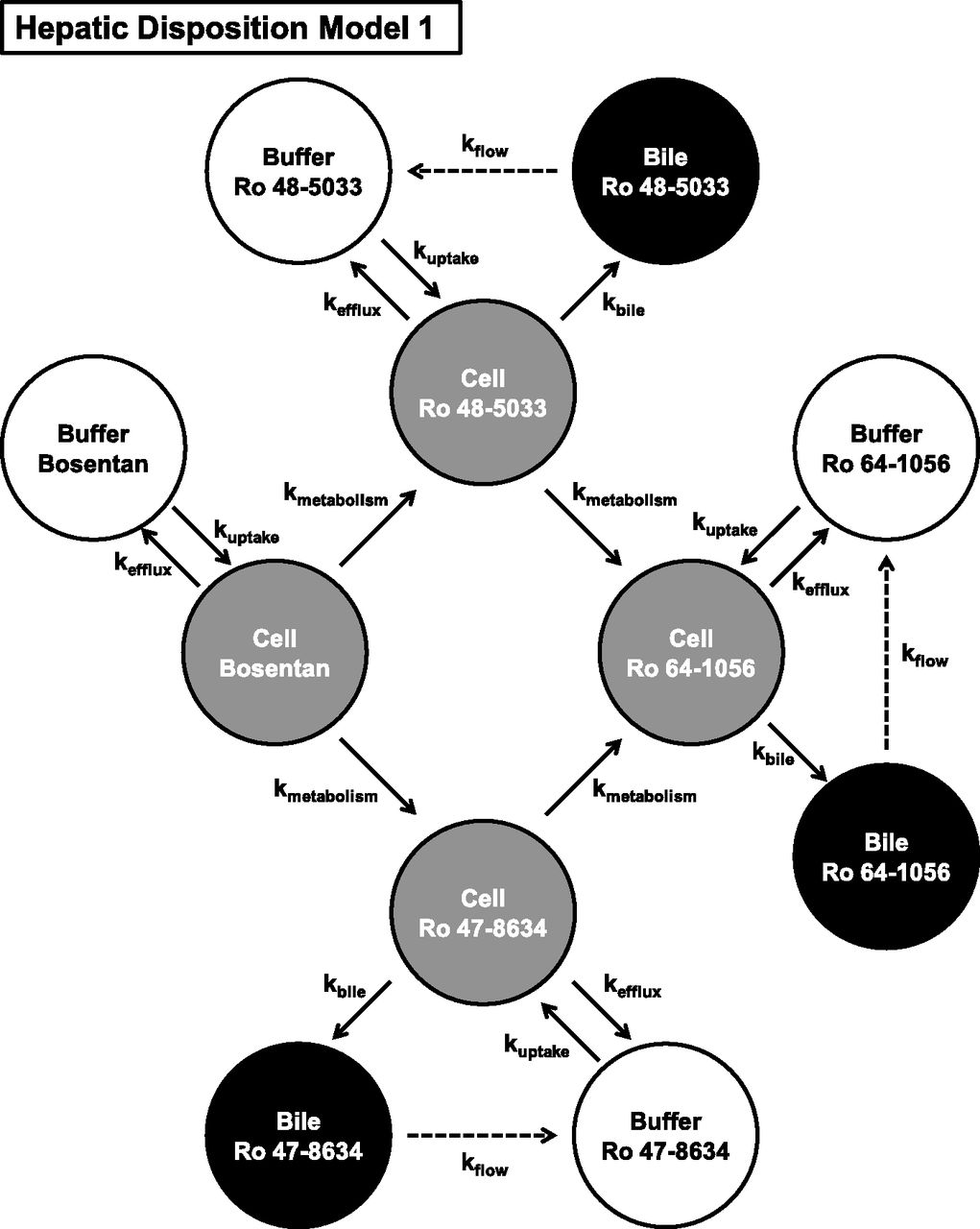
Hepatic disposition model of bosentan and its three metabolites in human SCHs based on the known metabolic pathway of bosentan.
Parameter estimates from the final population pharmacokinetic model
The parameters were estimated with NONMEM software (subroutine ADVAN 13) using the mass-time profiles of the hepatic disposition of bosentan and its three metabolites in human SCHs based on Fig. 3 (model 1) or Fig. 8 (model 2). Model 1 and model 2 represent a hepatic disposition model without or with a novel metabolic pathway of Ro 47-8634 to M4, respectively.

Diagnostic plots for observed versus population analysis–predicted values in the buffer, cells, and bile canalicular lumen of bosentan, Ro 48-5033, Ro 47-8634, and Ro 64-1056 based on the known metabolic pathway of bosentan (model 1). Correlations were examined between the predicted and observed values of bosentan in the buffer (A) and cells (B); Ro 48-5033 in the buffer (C), cells (D), and bile canalicular lumen (E); Ro 47-8634 in the buffer (F), cells (G), and bile canalicular lumen (H); and Ro 64-1056 in the buffer (I), cells (J), and bile canalicular lumen (K). The dashed lines represent a correlation coefficient of 1. In the model, most of the observed values are uniformly distributed around the unity line, whereas the intracellular masses of Ro 64-1056 are overestimated.
Novel Metabolic Pathway of Bosentan in the Human Liver.
To clarify the reason for the overestimation of intracellular Ro 64-1056, in vitro metabolism studies were performed using human liver microsomes. First, the metabolism of Ro 64-1056 was investigated using human liver microsomes with NADPH (P450 pathway) or UDPGA (uridine 5′-diphosphoglucuronosyltransferase (UGT) pathway). As shown in Fig. 5, A and B, Ro 64-1056 was hardly metabolized during 30 minutes of incubation with human liver microsomes in the presence of either NADPH or UDPGA. Second, the metabolism of Ro 48-5033 was examined. As shown in Fig. 5C, the formation of Ro 64-1056 was confirmed, but the metabolism of Ro 48-5033 was very slow in the reaction mixture of human liver microsomes with NADPH. Last, the metabolic pathway of Ro 47-8634 was similarly examined. As shown in Fig. 5D, Ro 47-8634 decreased and Ro 64-1056 was linearly formed in the reaction mixture of human liver microsomes with NADPH. In addition, during in vitro metabolism studies of Ro 47-8634, an unknown ion peak was observed with a retention time of around 6 minutes in MS chromatograms for the quantification of Ro 64-1056 with the same m/z (554.3 → 189.0), suggesting the presence of an additional metabolite. To determine the structure of the unidentified metabolite, we further analyzed the unknown ion peak by a product ion scan. As shown in Fig. 6A, Ro 64-1056 showed some specific MS/MS fragmentations, such as 189.3, 204.1, and 280.4. The unknown ion peak showed similar MS/MS fragmentations of 189.2 and 204.3, but an MS/MS fragmentation of 313.4 was also confirmed instead of 280.4 (Fig. 6B). The difference was studied for the MS/MS fragmentation patterns between Ro 64-1056 and the unknown ion peak, and the structure of the novel metabolite was determined as a hydroxylated form at any position of the phenol group of Ro 47-8634. It was found as a novel metabolite of bosentan and termed here as M4.

Concentration-time profiles of bosentan’s metabolites from in vitro metabolism studies. (A) Ro 64-1056 (0.5 µM) was incubated with human liver microsomes (0.5 mg/ml) in the presence of 1 mM NADPH for 0, 5, 10, 20, or 30 minutes. (B) Ro 64-1056 (0.5 µM) was incubated with 0.5 mg/ml human liver microsomes, which had been pretreated with 25 µg/ml alamethicin, in the presence of 2 mM UDPGA for 30 minutes. (C) Ro 48-5033 (0.5 µM) was incubated with human liver microsomes (0.5 mg/ml) in the presence of 1 mM NADPH for 30 minutes. Closed circles represent Ro 48-5033 concentrations, open circles represent formed Ro 64-1056 concentrations. (D) Ro 47-8634 (0.5 µM) was incubated with human liver microsomes (0.5 mg/ml) in the presence of 1 mM NADPH for 30 minutes. Closed circles represent Ro 47-8634 concentrations, open circles represent formed Ro 64-1056 concentrations. Data represent means ± S.D. from three independent experiments on human liver microsomes. UGT, uridine 5′-diphosphoglucuronosyltransferase.

MS/MS fragmentations by product ion scans. MS chromatograms and fragmentation patterns of Ro 64-1056 (A) and M4 (B). TIC, total ion current. To improve the resolution of the numerals on the X and Y axes, the traced figure of the original is shown.
Re-establishment of the Hepatic Disposition Model.
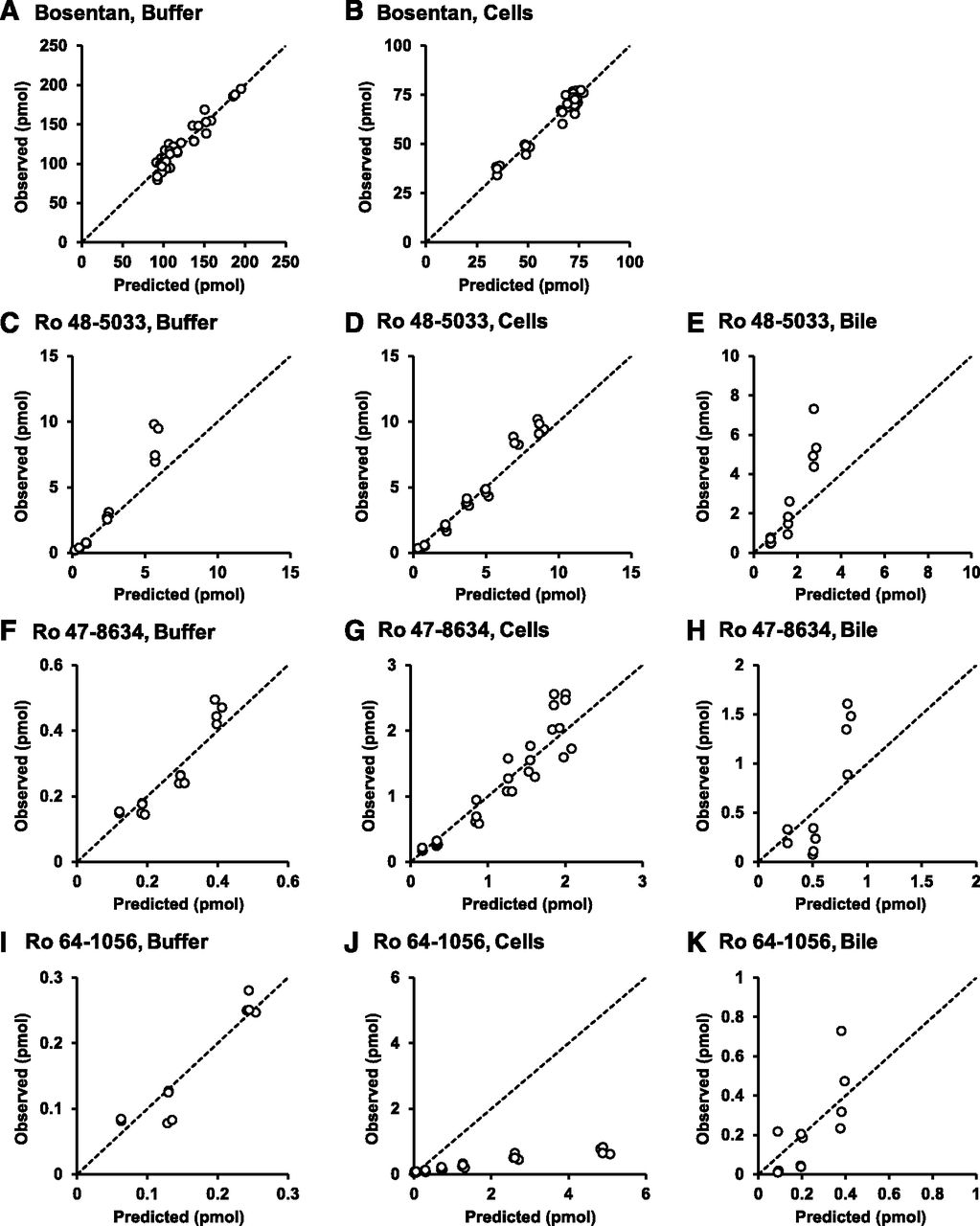
Figure 7 shows a newly proposed metabolic pathway of bosentan. The revised hepatic disposition model 2 incorporated the novel metabolic pathway of Ro 47-8634 to M4 to re-estimate the parameters of the first-order rate constants for basolateral uptake and efflux, intracellular metabolism, biliary excretion, and flow from the bile canalicular lumen to the buffer (Fig. 8). Figure 9 shows the correlations between the predicted and observed values based on the modified model. Significant improvement was observed in the predicted values of Ro 64-1056 in the cell compartment (Fig. 9J), without significant effects on the other values. The relative standard error of the estimate was below 30% for most of the pharmacokinetic parameters, suggesting adequate precision for these estimates. A final population pharmacokinetic model was successfully constructed, as shown in Table 2.

Proposed metabolic pathway of bosentan in the human liver by quantitative model analysis.

Hepatic disposition model of bosentan and its three metabolites in human SCHs with the metabolic pathway of Ro 47-8634 to M4.
Diagnostic plots for observed versus population analysis–predicted values in the buffer, cells, and bile canalicular lumen of bosentan, Ro 48-5033, Ro 47-8634, and Ro 64-1056 based on the known metabolic pathway of bosentan using the modified model incorporating the metabolic pathway of Ro 47-8634 to M4 (model 2). Correlations were examined between the predicted and observed values of bosentan in the buffer (A) and cells (B); Ro 48-5033 in the buffer (C), cells (D), and bile canalicular lumen (E); Ro 47-8634 in the buffer (F), cells (G), and bile canalicular lumen (H); and Ro 64-1056 in the buffer (I), cells (J), and bile canalicular lumen (K). The dashed lines represent a correlation coefficient of 1. Significant improvement was observed in the predicted values of Ro 64-1056 in the cell compartment, without significant effects on the other values.
Hepatotoxicity of Bosentan’s Metabolites.
For the study of the toxic effect of bosentan’s metabolites, we focused on Ro 47-8634 and Ro 64-1056, since hepatotoxicity of Ro 48-5033 has been reported to be negligible. Because CYP3A4 and CYP2C9 are involved in hydroxylation at the t-butyl group of Ro 47-8634 (van Giersbergen et al., 2002), the cytotoxic effect of Ro 47-8634 toward human hepatocytes was examined after pretreatment with dimethylsulfoxide alone (0.1%; vehicle control), TA (10 µM; specific CYP2C9 inactivator), ETM (10 µM; specific CYP3A inactivator), or 1-ABT (1 mM; broad P450 inactivator). As shown in Fig. 10A, apparent hepatotoxicity of Ro 47-8634 was observed at 200 µM, and the effect on cell viability was ameliorated by pretreatment with TA (P < 0.05). Pretreatment with 1-ABT also prevented the cytotoxicity of Ro 47-8634, but without statistical significance (P = 0.1240). On the other hand, no effect of pretreatment of ETM was observed for Ro 47-8634–induced cytotoxicity. The hepatotoxic effect of Ro 64-1056 was also examined. As shown in Fig. 10B, Ro 64-1056 significantly decreased the cell viability at concentrations higher than 100 µM.

Cytotoxic effect of Ro 47-8634 or Ro 64-1056 on human hepatocytes. (A) Data are shown for human hepatocytes pretreated with 0.1% dimethylsulfoxide (vehicle control; filled bars), 10 µM TA (specific CYP2C9 inactivator; striped bars), 10 µM ETM (specific CYP3A inactivator; open bars), and 1 mM 1-ABT (broad P450 inactivator; dotted bars). The hepatotoxic effect of Ro 47-8634 at 200 µM disappears after pretreatment with TA (*P < 0.05, paired t test). Pretreatment with 1-ABT also tends to maintain the cell viability, but without statistical significance. (B) Ro 64-1056 shows concentration-dependent cytotoxicity. Ro 64-1056 significantly decreased the cell viability at concentrations higher than 100 µM (*P < 0.05 at 100 µM and ***P < 0.001 at 200 µM, Dunnett’s test). Data represent means + S.D. from four lots of human hepatocytes (DQB, GMX, Hu1437, and Hu8110).
Discussion
Bosentan is known to cause hepatotoxicity, but its mechanism has not been fully understood. The liver is a major organ that determines the fates of endogenous and exogenous compounds and their metabolites in the body; at the same time, the liver may suffer from damage caused by these components directly or indirectly. Thus, in vitro methods that are applicable for evaluation of the hepatic disposition and toxicological effects of drugs in humans are expected. To consider the effects of both drugs and their metabolites on the liver, a quantitative analysis of their disposition is required in three compartments: blood, liver, and bile. The SCH system is a unique in vitro method that mimics the in vivo situation of the liver by maintaining three compartments, extracellular medium, hepatocytes, and bile canalicular lumen, which correspond to blood, liver, and bile compartments, respectively. For quantitative evaluation, a model analysis of drug disposition in those three compartments was applied. Accordingly, in the present study, the hepatic disposition of bosentan and its three metabolites (Ro 48-5033, Ro 47-8634, and Ro 64-1056) in human SCHs and the hepatotoxicity by bosentan’s two metabolites (Ro 47-8634 and Ro 64-1056) toward human hepatocytes were evaluated. The findings in the present study are summarized.
First, in the present study, a novel metabolite of bosentan, M4, was identified in the human liver. The ion peak of M4 in MS chromatograms was observed in the analytical samples of human SCHs (Supplemental Fig. 1). In a human mass balance study of 14C-bosentan, some unknown metabolite peaks were observed in the radiochromatograms in feces and urine (Weber et al., 1999). Considering the elution order of bosentan and its metabolites, M4 corresponds to the unknown metabolite at a retention time of around 22 minutes in the previous study, although our LC-MS/MS analysis was performed under slightly different LC conditions. Because M4 is detected in the radiochromatogram in urine (Weber et al., 1999), it is expected to be exposed in the circulation. Ro 64-1056 is the second-largest systemic metabolite in humans, followed by Ro 48-5033 (Dingemanse and van Giersbergen, 2004), and the present results showed that the in vitro formation rate of M4 seemed to be greater than that of Ro 64-1056 from Ro 47-8634 (Fig. 6). These findings suggest that M4 is systemically exposed at a certain level in humans.
Second, the finding of a novel metabolite, M4, was based on the model analysis of the hepatic disposition of bosentan and its previously known metabolites. Apparent discrepancy between observations and model-based predictions of hepatic intracellular amount of Ro 64-1056 in human SCHs made this finding possible. Accordingly, a combination of an in vitro measurement of hepatic disposition in SCHs and a quantitative model analysis should be useful to clarify the events of hepatic disposition of drugs, including parent compound and formed metabolites. The observed values of bosentan’s metabolites in bile canalicular lumen were relatively variable, but the predicted values of metabolites in the bile compartment mostly fell within 2-fold of the unity line, representing a correlation coefficient of 1. In addition, no obvious tendency of under- or overprediction was observed except for intracellular Ro 64-1056 in Fig. 4J, suggesting the hepatic disposition model for bosentan and its three metabolites in human SCHs was adequately established. Recently, model analyses have become widespread in various research fields. During drug discovery and development, model-based studies have been widely accepted with quantitative analyses such as systems biology and pharmacokinetics–pharmacodynamics (Milligan et al., 2013; Kimko and Pinheiro, 2015). In addition, the application of physiologically based pharmacokinetics models has increased in various studies for simulating pharmacokinetics in special populations (Edginton and Willmann, 2008; Ke et al., 2012; Strougo et al., 2012), evaluating drug-drug interaction risks (Baneyx et al., 2014; Gertz et al., 2014; Zamek-Gliszczynski et al., 2014), and predicting tissue-specific disposition at the sites of drug action (Gallo, 2013; Ball et al., 2014). These model-based studies can be further advanced by incorporating in vitro data. Indeed, bile acid–mediated hepatotoxicity by troglitazone in humans was recently predicted by the combination of a physiologically based pharmacokinetics model and mechanistically based mathematical models integrated with in vitro data (Yang et al., 2014). Hence, modeling of in vitro hepatic disposition is useful for the development of further model-based analyses, and in vitro data, such as those obtained in the present study, are also expected to be incorporated into such advanced applications.
Third, in the present hepatocyte toxicity studies, specific CYP2C9 inhibition by pretreatment with TA did ameliorate the decrease in cell viability after exposure with Ro 47-8634, suggesting that CYP2C9-mediated metabolite(s) can show hepatotoxicity after addition of Ro 47-8634 to human hepatocytes. Therefore, Ro 47-8634 was incubated with human CYP2C9.1-, CYP2C9.2-, or CYP2C9.3-expressing supersomes to investigate the involvement of CYP2C9 in the formation of Ro 64-1056 and/or M4. As shown in Supplemental Fig. 2, formation of Ro 64-1056 was found in all three CYP2C9 genotype-expressing supersomes, whereas formation of M4 was not observed, meaning that CYP2C9 is specifically involved in hydroxylation of the t-butyl group of Ro 47-8634. The hepatotoxic effect of Ro 47-8634 also tended to disappear in human hepatocytes pretreated with 1-ABT, although the recovery was not statistically significant. This may arise through the weak inactivation potential of 1-ABT for CYP2C9 compared with other P450 isoforms (Kimoto et al., 2012). On the other hand, ETM-mediated specific CYP3A inhibition did not affect the cytotoxicity induced by Ro 47-8634. This may be due to the low contribution of CYP3A4 to the metabolism of Ro 47-8634 to Ro 64-1056, because CYP2C9 is primarily involved in hydroxylation at the (t-butyl group of bosentan) (Markova et al., 2013). These results suggest a negligible potential of Ro 47-8634 and M4 to cause hepatotoxicity, and a possible role of the formed Ro 64-1056 in the cytotoxicity after addition of Ro 47-8634 to human hepatocytes. Accordingly, the metabolic pathway of Ro 47-8634 to M4 might be an alternative to avoid Ro 64-1056–induced liver injury. Because the formation rate of M4 seemed to be higher than that of Ro 64-1056 from Ro 47-8634 in human liver microsomes (Fig. 6), if formation of M4 decreases in certain situations, it may lead to an increase in Ro 64-1056 formation. Therefore, it is important to identify which P450 isoforms are responsible for the metabolism from Ro 47-8634 to M4 in the future.
Although bosentan is well known as a hepatotoxic drug, the mechanism of its hepatotoxicity is still not fully understood. This is the first study to report that Ro 64-1056 has cytotoxic potential for human hepatocytes, and to indicate the possibility that Ro 64-1056 is involved in bosentan-induced liver injury, at least in part. Markova et al. (2013) recently proposed CYP2C9*2 as a potential genetic marker for bosentan-induced liver injury, indicating the possible association of decrease of CYP2C9 activity with bosentan-induced liver injury. This clinical information is inconsistent with our present results, because the genetic polymorphisms of CYP2C9 can decrease Ro 64-1056 formation from Ro 47-8634 in the liver; however, bosentan itself shows hepatotoxicity (Markova et al., 2013), and the decrease of CYP2C9 activity can also lead to continuous exposure of bosentan in the liver. In addition, the exact genetic impact of CYP2C9 is still vague (Roustit et al., 2014). Therefore, further clinical studies may be required to confirm the genetic impacts on bosentan-induced liver injury. Drug-induced liver injury is proposed to be linked to certain primary mechanisms, including direct cytotoxicity, reactive metabolite formation, inhibition of BSEP, and mitochondrial dysfunction (Aleo et al., 2014). In the present in vitro metabolism studies, Ro 64-1056 was minimally metabolized in human liver microsomes, and therefore, the reactive metabolite formation is unlikely to be involved. Cholestasis is thought to be one of the hepatotoxic mechanisms of bosentan, and the canalicular efflux transporter BSEP and/or sinusoidal efflux transporters multidrug resistance-associated proteins 3 and 4 play roles in intracellular bile acid homeostasis (Fattinger et al., 2001; Zollner et al., 2007; Vanwijngaerden et al., 2011; Dawson et al., 2012; Treiber et al., 2014). In addition, disruption of mitochondrial function can lead to oxidative stress, apoptosis, and hepatocellular injury along with a reduction of intracellular ATP levels, and the combination of mitochondrial and BSEP inhibition should be more highly associated with the severity of human drug-induced liver injury (Aleo et al., 2014). Bosentan-induced liver injury was underpredicted using a mechanistic model linked with the inhibition constants of bosentan (12 µM) and Ro 47-8634 (8.5 µM) for rat Bsep (Woodhead et al., 2014). Concentrations of Ro 64-1056 showing significant cytotoxicity for human hepatocytes were relatively high, but this discrepancy may be due to Ro 64-1056 not being a good substrate for hepatic uptake transporters and/or poor permeability of Ro 64-1056, because hydrophilic metabolites usually show less membrane permeability than their lipophilic parent compounds. Therefore, the results of hepatotoxicity studies should be carefully considered, especially using metabolites; however, our present results indicate that Ro 64-1056 is involved in bosentan-induced liver injury, at least in part. In addition, among the metabolites of bosentan, Ro 48-5033 seemed to be a substrate for the biliary excretion transporters multidrug resistance-associated protein 2 and breast cancer resistance protein in vesicular transport studies using human canalicular transporter–expressing membrane vesicles (Supplemental Fig. 3). Because the effects of bosentan’s metabolites on mitochondrial function and/or these transporters involved in the hepatic disposition of bile acid in humans have not been fully evaluated, further experiments may be warranted to investigate their association with bosentan-induced liver injury.
In conclusion, we identified a novel metabolic pathway of Ro 47-8634 to M4 in the human liver by a quantitative model analysis, and succeeded in establishing a hepatic disposition model of bosentan and its metabolites in human SCHs. In addition, Ro 64-1056 exhibited a concentration-dependent hepatotoxic potential, and the metabolism of Ro 47-8634 to Ro 64-1056 by CYP2C9 was essential for Ro 64-1056–induced hepatotoxicity, whereas CYP2C9 was not involved in M4 formation. Therefore, the novel metabolic pathway identified can be considered as an alternative route that can avoid hepatotoxicity of Ro 64-1056. The underlying mechanism for the hepatotoxicity of Ro 64-1056 can be a clue toward better understanding of bosentan-induced liver injury.
Acknowledgments
The authors thank Tamami Arai for help with preparing the manuscript. The authors also appreciate Junsaku Kitagawa and Dr. Chihiro Hasegawa for their assistance in constructing the hepatic disposition models.
Authorship Contributions
Participated in research design: Matsunaga, Nakanishi, Nunoya, Imawaka, Tamai.
Conducted experiments: Matsunaga, Kaneko, Staub.
Performed data analysis: Matsunaga, Kaneko.
Wrote or contributed to the writing of the manuscript: Matsunaga, Kaneko, Staub, Nakanishi, Nunoya, Imawaka, Tamai.
Footnotes
- Received September 3, 2015.
- Accepted October 23, 2015.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- 1-ABT
- 1-aminobenzotriazole
- BSEP
- bile salt export pump
- ETM
- erythromycin
- LC-MS/MS
- liquid chromatography with tandem mass spectrometry
- P450
- cytochrome P450
- SCH
- sandwich-cultured hepatocyte
- TA
- tienilic acid
- UDPGA
- uridine 5′-diphosphoglucuronic acid
- WME
- Williams’ medium E
- WST-8
- 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}