


Abstract
The prodrug clopidogrel (Plavix) is activated by cytochrome P450 (P450) to a metabolite that inhibits ADP-induced platelet aggregation. Clopidogrel is frequently administered to patients in conjunction with the CYP3A4 substrate atorvastatin (Lipitor). Since clinical studies indicate that atorvastatin inhibits the antiplatelet activity of clopidogrel, we investigated whether CYP3A4 metabolized clopidogrel in vitro. Microsomes prepared from dexamethasone-pretreated rats metabolized clopidogrel at a rate of 3.8 nmol min−1nmol of P450−1, which is 65 and 1270% faster than the rate of metabolism by microsomes from control and β-napthoflavone-treated rats, respectively. To identify the human P450s responsible for clopidogrel oxidation, genetically engineered microsomes containing a single human P450 isozyme were tested for their ability to oxidize clopidogrel. CYP3A4 and 3A5 metabolized clopidogrel at a significantly higher rate than eight other P450 isozymes, suggesting that CYP3A4 and 3A5 are primarily responsible for in vivo clopidogrel metabolism. Clopidogrel interacts with human CYP3A4 with a spectral dissociation constant (Ks),Km, and Vmax of 12 μM, 14 ± 1 μM and 6.7 ± 1 nmol min−1nmol P450−1, respectively. Atorvastatin lactone, the physiologically relevant substrate, inhibits clopidogrel with aKi of 6 μM. When clopidogrel and atorvastatin are present at equimolar concentrations, clopidogrel metabolism is inhibited by greater than 90%. Since CYP3A4 and 3A5 metabolize clopidogrel faster than other human P450 isozymes and are the most abundant P450s in human liver, they are predicted to be predominantly responsible for the activation of clopidogrel in vivo.
Clopidogrel hydrogen sulfate (d-methyl[2-chlorophenyl]-5-[4,5,6,7- tetrahydrothieno] [3,2-c pyridinyl] acetate hydrogensulfate) is a thienopyridine prodrug used clinically to inhibit ADP-induced platelet aggregation (Quinn and Fitzgerald, 1999). It reduces the risk of thrombotic events in patients with a history of artherosclerotic diseases, such as stroke or myocardial infarction (Coukell and Markham, 1997; Bertrand et al., 2000). In a 19,185 patient trial, it was demonstrated that clopidogrel was superior to aspirin in lowering the occurrence of ischemic events from 5.83% in patients using aspirin to 5.32% in patients using clopidogrel (CAPRIE Steering Committee, 1996). Clopidogrel was particularly effective at reducing the occurrence of ischemic events in patients having a history of cardiac surgery with a relative risk reduction of 22% when compared with aspirin (Bhatt et al., 2000).
Clopidogrel requires oxidation by hepatic cytochrome P450 (P4501) to generate a metabolite that is an active inhibitor of ADP-induced platelet aggregation (Savi et al., 1994). However, only a small proportion of administered clopidogrel is metabolized by P450. The majority of clopidogrel is hydrolyzed by esterases to an inactive carboxylic acid derivative that accounts for 85% of the clopidogrel-related compounds circulating in plasma. Clopidogrel itself is not detected in plasma (Reist et al., 2000). P450 catalyzes the oxidation of the thiophene ring of clopidogrel to 2-oxoclopidogrel. The 2-oxo-intermediate is then oxidized further by P450. The second oxidation results in opening of the thiophene ring to form both a carboxyl and a thiol group (Savi et al., 2000). The thiol group forms a disulfide bond with the P2Y12ADP-receptor on platelets. ADP cannot bind to the covalently modified receptor, which normally activates the glycoprotein GPIIb/IIIa complex that binds fibrinogen thereby initiating clot formation (Savi et al., 2001).
Clopidogrel is structurally similar to other thienopyridines including ticlopidine, tienilic acid, and CS-747. These compounds have been extensively characterized and are also metabolized by P450. Ticlopidine, which is metabolized by human CYP2C19, is also a suicide inhibitor of CYP2C19 and a known competitive inhibitor of CYP2D6 (Ha-Duong et al., 2001). Tienilic acid is both oxidized by, and a suicide inhibitor of, human CYP2C9 (López-Garcia et al., 1994). The third thienopyridine, CS-747, is oxidized by human CYP3A4 and 2B6 (Kazui et al., 2001). The human P450 responsible for oxidation of clopidogrel has not been identified, but it has been suggested that clopidogrel is oxidized by rat CYP1A2 (Savi et al., 1994).
Atorvastatin, the most widely prescribed pharmaceutical for the prevention of hypercholesterolemia, is a member of the “statin” family. The statins are a group of HMG-CoA reductase inhibitors that inhibit cholesterol biosynthesis by binding to the active site of HMG-CoA reductase (Igel et al., 2001). Atorvastatin is administered as the calcium salt of atorvastatin acid and is rapidly converted to its lactone form by a number of enzymatic processes, including acyl CoA-synthase (Prueksaritanont et al., 2001), paraoxonase (Teiber et al., 2002), and glucuronosyltransferases (Prueksaritanont et al., 2002). Conversely, the lactone form of atorvastatin undergoes hydrolysis back to atorvastatin acid through the action of esterases. Clinical studies have demonstrated that the serum concentration of atorvastatin acid and lactone are similar in vivo (Kantola et al., 1998), indicating that the two forms of atorvastatin are in dynamic equilibrium in vivo. Both forms of atorvastatin are hydroxylated by CYP3A4, but the Km for atorvastatin lactone hydroxylation is 1.4 μM, ∼20 times lower than theKm of 25 μM for atorvastatin acid hydroxylation. The greater affinity of atorvastatin lactone for CYP3A4 indicates that the primary route of atorvastatin metabolism in humans is via the lactone (Jacobsen et al., 2000).
Clinical studies have demonstrated that atorvastatin inhibits the antiplatelet activity of clopidogrel (Lau et al., 2000). More recently, the anti-platelet activity of clopidogrel was observed to be inhibited in vivo by the CYP3A4 inhibitors erythromycin and troleandomycin and stimulated by the CYP3A4 inducer, rifampin (Lau et al., 2002). In this study, in vitro experiments have been performed in an attempt to unambiguously identify the human P450s responsible for clopidogrel oxidation.
Experimental Procedures
Materials.
Atorvastatin was generously provided by Pfizer (Groton, CT). 7-Ethoxyresorufin was purchased from Molecular Probes (Eugene, OR). Erythromycin was from ICN Biomedicals (Aurora, OH). HPLC grade acetonitrile and ammonium acetate were from Fisher Scientific Co. (Fair Lawn, NJ). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Rat liver microsomes were a gift from Paul Hollenberg and were isolated from the livers of fasted male Fischer rats (175–190 g; Harlan Sprague-Dawley, Indianapolis, IN) that had been treated with either dexamethasone or β-napthoflavone (Chun et al., 2000).
Microsomes prepared from Spodoptera frugiperda insect cells that had been infected with a baculovirus containing the cDNA coding for P450 reductase and a single human P450 isozyme were purchased from two different sources. Insect cell microsomes containing CYP1A2, 2C9, 2C19, 2D6, 3A4, or 3A5 were purchased from PanVera (Madison, WI) whereas insect cell microsomes containing CYP1A1, 2A6, 2E1, or 2B6 were purchased from BD Gentest (Woburn, MA) (Lee et al., 1996).
Microsomes prepared from human B lymphoblasts that had been transformed with a vector derived from the pHEBo vector, which contains the Epstein Barr OriP sequence, an Escherichia coli HisD gene, plasmids pBR322 and pHSV106, and the cDNA for either human CYP1A2, 2C9, or 3A4, were purchased from BD Gentest (Crespi, 1991).
The cytochrome c reductase activity of the purchased microsomes was used to determine the approximate concentration of P450 reductase by assuming that 1 nmol of pure, fully active P450 reductase reduced 4.47 μmol of cytochrome cmin−1 (Shen and Kasper, 2000). Insect cell microsomes typically contained P450/P450 reductase at a molar ratio of 1:0.2–7, whereas human B lymphoblast microsomes contained P450/P450 reductase at a molar ratio of 1:0.05–0.2. All microsomes were stored as aliquots at −80°C prior to use. The membrane bound form of rabbit cytochrome b5 was prepared as described by Mulrooney and Waskell (2000).
Measurement of Clopidogrel Oxidation.
The rate of P450-dependent clopidogrel oxidation was essentially measured as previously described (Savi et al., 1994) except that the reaction was conducted in the presence of 5 mM glutathione. Microsomes were gently shaken in reaction mixtures containing 50 μM clopidogrel, 1% methanol (a solvent for the clopidogrel), 100 mM potassium phosphate, pH 7.4, 100 mM KF (to inhibit esterase activity), 5 mM reduced glutathione, and 1 mM NADPH-regenerating mixture (1 mM NADP+, 1 mM glucose 6-phosphate, 1 U/ml glucose-6–phosphate dehydrogenase). The concentration of microsomal P450 used in the reaction mixture varied depending on the expression system. Reaction mixtures containing microsomes prepared from rat liver contained 1.0 μM P450. Reactions performed with microsomes from transformed human lymphoblasts contained 80 to 100 nM P450, and reaction mixtures using microsomes from S. frugiperda insect cells contained 30 to 50 nM.
To measure the rate of clopidogrel oxidation, reaction mixtures were incubated at 37°C for 5 min before addition of the NADPH-regenerating mixture to initiate the reaction. Aliquots (200 μl) were removed at timed intervals and mixed with 100 μl of ice-cold acetonitrile to stop the reaction. Aliquots were then centrifuged at 10,000 rpm for 10 min and stored at −80°C prior to HPLC analysis. Pseudo-first order rate constants for clopidogrel oxidation were determined using the following rate equation:
The amount of clopidogrel metabolized by individual human cytochrome P450 isozymes in insect cell microsomes was measured using 200 μl of reaction mixtures to minimize the P450 required. The reaction mixtures were incubated at 37°C for 5 min prior to the addition of microsomes to a final concentration of 30 nM. The reaction was allowed to proceed for 10 min. Ice-cold acetonitrile (100 μl) was added to the reaction mixtures, which were centrifuged and stored at −80°C before the remaining clopidogrel was measured using HPLC.
Experiments to measure the inhibition of clopidogrel oxidation by atorvastatin were performed using 200 μl of reaction mixtures prepared as above except that the reaction mixtures contained either 0 to 500 μM atorvastatin acid or 0 to 43 μM atorvastatin lactone. The reaction was started by adding human B lymphoblast microsomes containing cytochrome CYP3A4 or 3A5 to a final P450 concentration of 90 nM. The inhibition constant Ki was calculated using SigmaPlot (SPSS Science Inc., Chicago, IL)
To determine the effect of cytochromeb5 (cytb5) on the rate of clopidogrel oxidation, insect cell microsomes containing 2 μM CYP3A4 and 0.29 μM P450 reductase were incubated with 0 to 4 μM of the membrane bound form of cyt b5 for 30 min at 37°C to allow cyt b5 to enter the microsomal membrane (Vergeres et al., 1995). These microsomes were then added to reaction mixtures to give a final concentration of 100 nM CYP3A4, 14 nM P450 reductase, and 0 to 200 nM cytb5. The reaction mixtures were incubated for 30 min at 37°C, and the remaining clopidogrel was measured using HPLC.
The effect of CO on clopidogrel metabolism was investigated by passing CO over the surface of reaction mixtures containing microsomes for 5 min. The reaction was started by the addition of NADPH and gently agitated for 30 min before measuring the amount of clopidogrel metabolized.
Detection of Clopidogrel Using Reverse-Phase HPLC.
It was necessary to measure clopidogrel oxidation by the disappearance of clopidogrel rather than the production of metabolite due to the poor solubility of clopidogrel in aqueous solvents and the difficulty in accurately quantifying the products of clopidogrel oxidation. High performance liquid chromatography was performed on a PerkinElmer LC 450 HPLC equipped with an LC-252 detector. Two hundred microliters of the supernatant from the centrifuged mixture were injected onto a Waters RESOLVE C18 column (15 cm × 4.6 mm, 5-μm particle size; Waters, Milford, MA). Solvent A was distilled water containing 10 mM ammonium acetate and 10% acetonitrile whereas solvent B was acetonitrile. A linear gradient of 17 to 43% solvent B for 30 min, then 43 to 100% B over 15 min was used to elute the components of the reaction mixture. Clopidogrel was eluted after 26 min at a flow rate of 1 ml min−1 and detected by UV-visible spectrophotometry at 220 nm where clopidogrel has an absorption coefficient of 12000 M−1cm−1. This peak was identified as clopidogrel using a VG Fisons “Platform” single quadrupole electrospray mass spectrometer (Micromass, Inc., Beverly, MA). The mass spectrum of clopidogrel exhibited a protonated ion at m/z322.2. The amount of clopidogrel was quantified by integration of the HPLC peak corresponding to clopidogrel and comparison to a standard curve.
Determination of the Spectral Dissociation Constant (Ks) between Clopidogrel and Human Cytochrome P450.
The Ks between clopidogrel and CYP3A4 was determined using UV-visible spectroscopy. Substrate binding to the putative recognition site on P450 causes a change in the spin-state of the ferric heme group from low-spin hexacoordinate to high-spin pentacoordinate due to a displacement of the water molecule from the distal 6th ligand position of the heme. The change in spin state can be measured as a type 1 spectral change in the Soret band of the P450 spectrum, which is characterized by an increase in absorbance at 385 nm and a decrease at 420 nm (Sligar, 1976).
Reaction mixtures containing P450 (0.1 μM) in insect cell microsomes, 100 mM KF, 100 mM potassium phosphate, pH 7.4, and 20% glycerol were equilibrated at 37°C. Aliquots of clopidogrel were added to the incubation mixture and equilibrated for a further 2 min. The spectrum was recorded between 300 and 700 nm on a CARY Bio 300 UV-visible spectrophotometer (Varian Inc. Palo Alto, CA) and the difference in absorbance between 385 and 420 nm was measured. A double reciprocal plot was used to determine the Ksbetween CYP3A4 and clopidogrel.
Measurement of 7-Ethoxyresorufin Deethylase Activity.
Rat microsomal CYP1A2 activity was measured using the CYP1A2 specific substrate ethoxyresorufin as described previously (Kent, 1998). Reaction mixtures (1 ml) containing 5 μM 7-ethoxyresorufin in 50 mM Tris-HCl, pH 7.4, 50 mM MgCl2, and 1 mM NADPH-regenerating mixture were equilibrated at 37°C for 5 min at which time microsomes were added to a final P450 concentration of 0.2 μM. The reaction mixtures were gently shaken for 30 min at 37°C. The reaction was stopped by adding 750 μl of ice-cold methanol. The amount of resorufin product was measured by the increase in fluorescence on a Shimadzu RF-5000U spectrofluorophotometer (Shimadzu Scientific Instruments Inc., Columbia, MD) with excitation at 522 nm and emission at 585 nm. Resorufin was quantified by comparison with a standard curve.
Measurement of Erythromycin N-Demethylation.
The N-demethylation rate of erythromycin, a specific substrate for CYP3A4, was determined by quantifying the amount of formaldehyde product as previously described (Kent et al., 1998). Reaction mixtures (0.5 ml) containing 100 μM erythromycin, 50 mM potassium phosphate, pH 7.4, 30 mM MgCl2, and 1 mM NADPH-regenerating mixture were equilibrated at 37°C for 5 min. Microsomes containing P450 (0.2 nmol) were then added, and the reaction mixtures were gently shaken for 15 min at 37°C. The reaction was terminated with 250 μl of 60% trichloroacetic acid. The amount of formaldehyde generated was measured by adding 250 μl of NASH's reagent (Nash, 1953) to the assays and incubating for 15 min at 57°C. Detection by fluorescence with excitation at 410 nm and emission at 510 nm was used to measure the amount of formaldehyde formed.
Measurement of Testosterone 6β-Hydroxylation.
Testosterone 6β-hydroxylation is specific for CYP3A4 and was measured as described previously (Ding and Coon, 1994). Reaction mixtures (0.5 ml) containing 50 mM HEPES buffer, pH 7.5, 7.5 mM MgCl2, 0.4 mM testosterone, and 0.4 mM NADPH were equilibrated for 5 min at 37°C. The reaction was initiated by addition of microsomes containing P450 at a final concentration of 0.1 μM. Reaction mixtures were incubated for 20 min at 37°C before the addition of 1 ml of ethyl acetate. The ethyl acetate phase was removed and dried under a nitrogen stream. The residue containing the metabolites was dissolved in 110 μl of 65% methanol, and the amount of 6β-hydroxytestosterone formed was measured using isocratic HPLC (Rainin 25 × 4.9 mm; 5-μm C18 column; mobile phase, 35% methanol; Rainin Instrument Co. Inc. Woburn, MA) and quantified at 254 nm using 6β-hydroxytestosterone as a standard.
Synthesis of Atorvastatin Lactone.
Atorvastatin calcium (4 mM) was incubated in 1 mM HCl, pH 3.0, for 3 h at 80°C. After 3 h, acetonitrile was added to a final concentration of 33%, and this mixture was injected into the HPLC where the acid and lactone forms were completely resolved. The acid and lactone containing fractions were collected separately as described byKearney et al. (1993). The isolated lactone was dried by vacuum centrifugation and dissolved in methanol. The identity of the purified acid and lactone forms of atorvastatin was verified using a VG Fisons Platform single quadrupole mass spectrometer equipped with an electrospray ionization source. Protonated molecular ions were observed for the acid and the lactone at m/z 559.3 and 541.4, respectively.
Results
Metabolism of Clopidogrel by Rat Liver Microsomes.
The ability of rat liver microsomes to metabolize clopidogrel was determined by measuring the amount of clopidogrel remaining after incubation of clopidogrel in the presence of hepatic microsomes that had been prepared from either control rats or rats that had been pretreated with dexamethasone or β-napthoflavone as described underExperimental Procedures. The concentration of clopidogrel remaining was quantified by comparison to a standard curve, and the amount of clopidogrel metabolized was measured by subtracting the remaining clopidogrel from the initial amount of clopidogrel (Fig.1).
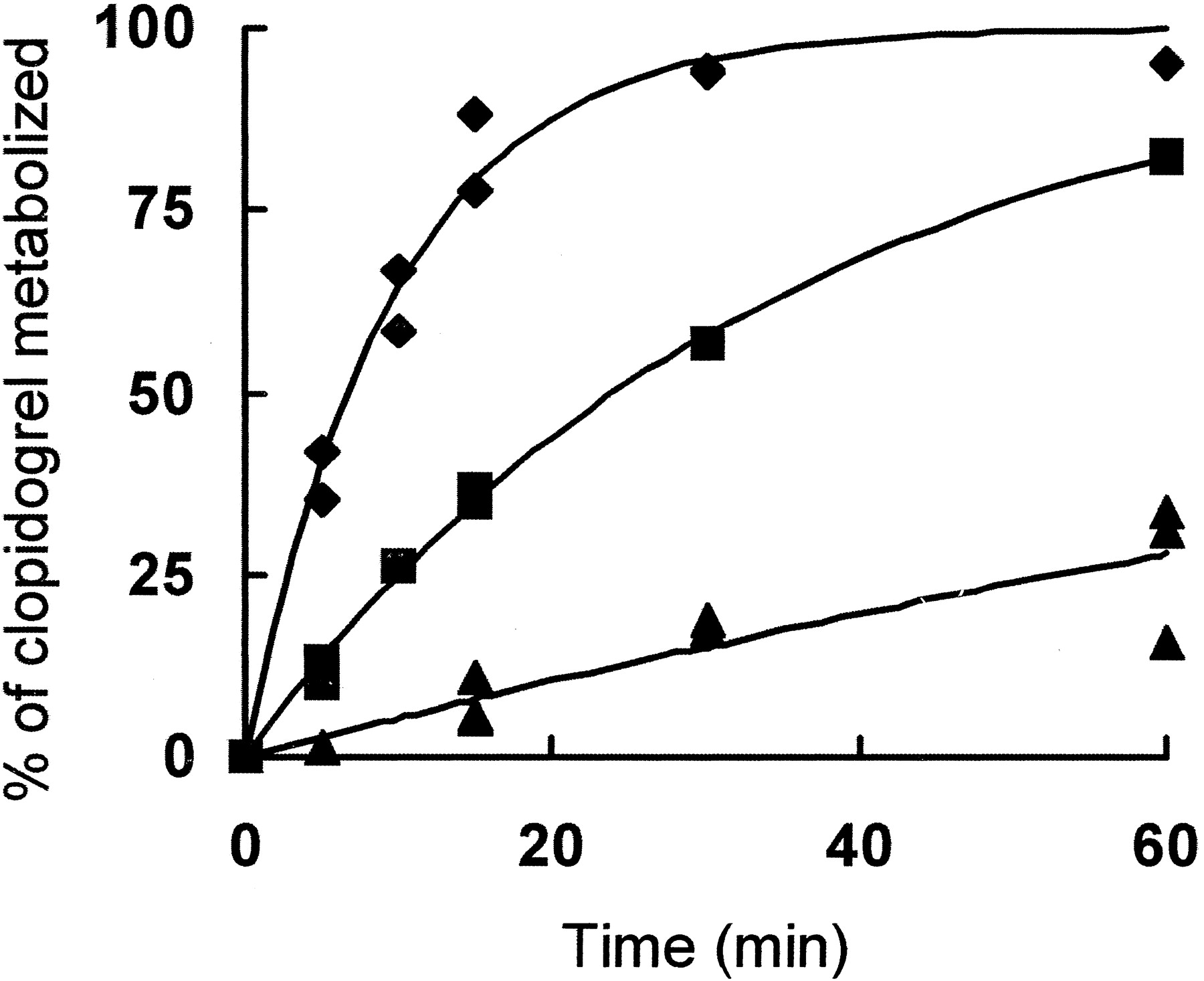
Oxidation of clopidogrel by rat liver microsomes.
Clopidogrel (50 μM) was incubated with 0.5 to 1 μM P450 in liver microsomes from control rats (0.5 μM P450, ▪) or rats treated with β-napthoflavone (1 μM P450, ▴) or dexamethasone (1 μM P450, ♦) as described under Experimental Procedures. At the indicated times, aliquots were removed, and the remaining clopidogrel was quantified.
Hepatic microsomes prepared from rats treated with dexamethasone have increased levels of CYP3A, whereas microsomes from rats treated with β-napthoflavone have increased levels of CYP1A1 and 1A2 (Wortelboer et al., 1991). The initial rate of clopidogrel metabolism by liver microsomes from control rats or rats treated with dexamethasone or β-napthoflavone was 2.3, 3.9, and 0.3 nmol of clopidogrel metabolized min−1 nmol P450−1, respectively. The data were also fitted using pseudo first-order rate constants and similar values (2.9, 5.1, and 0.3 min−1 for control, dexamethasone-induced and β-napthoflavone-induced microsomes, respectively) were obtained. Control experiments demonstrated that clopidogrel metabolism was not observed in the absence of NADPH and was inhibited by CO.
Erythromycin N-demethylation and testosterone 6β-hydroxylation are catalyzed almost exclusively by CYP3A in rats whereas 7-ethoxyresorufin deethylation is only catalyzed by CYP1A2 (Kent et al., 1998). These substrates were used to assess the activity of CYP3A and 1A2 in microsomes of rats pretreated with dexamethasone and β-napthoflavone, respectively (Table1). The rate of clopidogrel oxidation and the rate of both testosterone hydroxylation and erythromycin demethylation were highest in hepatic microsomes with the highest concentration of CYP3A, prepared from the livers of rats pretreated with dexamethasone. Liver microsomes prepared from rats treated with β-napthoflavone had the lowest rate of testosterone hydroxylation, erythromycin demethylation, and clopidogrel oxidation. The rate of clopidogrel metabolism was directly proportional to the rate of other CYP3A substrates suggesting that clopidogrel is a good substrate of CYP3A in rats.
Substrate metabolism by rat liver microsomes
In contrast, the rate of 7-ethoxyresorufin deethylation was highest in β-napthoflavone-induced microsomes, which contain high levels of CYP1A2, and lowest in microsomes from dexamethasone pretreated rats, which contain high levels of CYP3A (Wortelboer et al., 1991). These data demonstrated that clopidogrel is metabolized most effectively by rat liver microsomes containing increased levels of CYP3A, whereas rat liver microsomes containing increased levels of CYP1A2 metabolize clopidogrel at much lower rates than microsomes obtained from control rats.
Identification of the Human P450 Isozymes Responsible for Clopidogrel Oxidation.
Table 2 shows the average rate of clopidogrel oxidation by a single P450 isozyme expressed in insect cell microsomes, which have a P450/P450 reductase ratio of ≅ 1:3–5. A major limitation of this approach was the difficulty in accurately measuring low rates of clopidogrel metabolism due to the poor signal to noise ratio. The amount of clopidogrel remaining in the reaction mixture after incubation with CYP1A1, 2A6, 2C9, 2C19, 2D6, and 2E1 was not statistically significantly different from the amount of clopidogrel present at the beginning of the reaction. These data were interpreted to indicate that these six isozymes did not metabolize clopidogrel. CYP1A2 and 2B6 metabolized a low but statistically significant amount of clopidogrel whereas CYP3A4 and 3A5 clearly metabolized a significant amount of clopidogrel. Carbon monoxide inhibited greater than 90% of clopidogrel metabolism, which rules out the participation of flavin monoamine oxidase in clopidogrel oxidation.
Clopidogrel metabolism by different human P450 isozymes expressed in insect cell microsomes
In general, substrates are metabolized ≅ 7- to 10-fold more rapidly in insect microsomes than in either human liver or lymphoblast microsomes because of their high level of reductase relative to P450. The rate of substrate metabolism is comparable in human liver and lymphoblast microsomes. An advantage of microsomes from lymphoblasts and insect cells is that they contain only a single isozyme of P450. Thus, they are more useful than human liver microsomes when one is attempting to identify which P450 isozyme metabolizes a particular substrate. Although the absolute rates of metabolism are greater in insect cell microsomes, the relative rates of substrate metabolism are expected to be the same in both systems (Crespi, 1991; Shaw et al., 1997).
Clopidogrel metabolism was also investigated in microsomes from human Β lymphoblasts, which had been transformed with the cDNA of either CYP3A4, 2C9 or 1A2. Figure 2 illustrates that CYP3A4 metabolizes clopidogrel at rates of 90 and 4 nmol min−1 nmol−1 P450 in insect cell and lymphoblast microsomes, respectively, whereas clopidogrel essentially is not metabolized in either system by CYP1A2. CYP2C9 did not catalyze detectable amounts of clopidogrel metabolism. The rate of clopidogrel metabolism in human lymphoblast microsomes is similar to that in rat microsomes from dexamethasone-treated animals (Table 1).
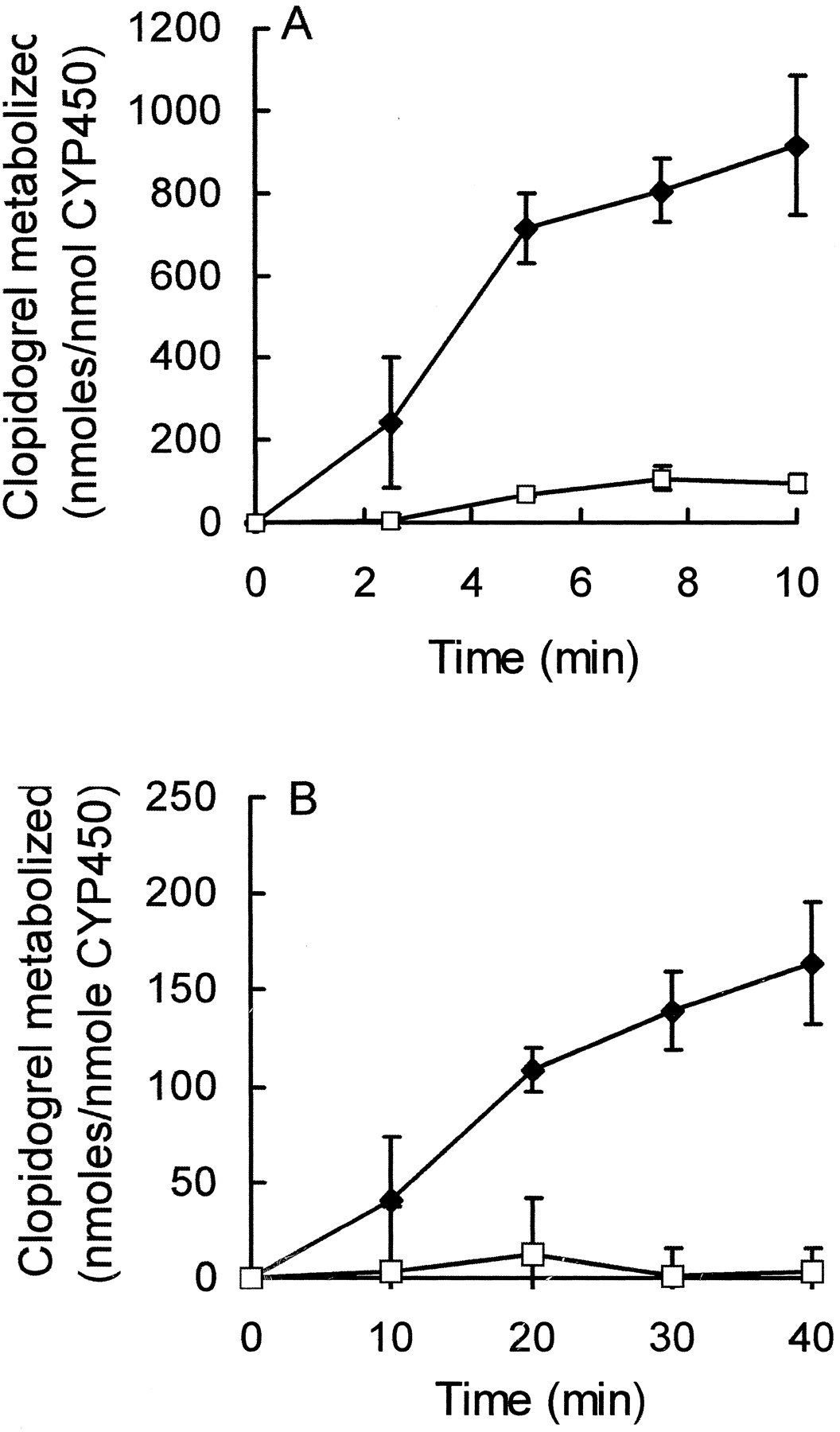
Comparison of clopidogrel oxidation by CYP3A4 and 1A2 expressed in either insect cell or human B lymphoblast microsomes.
A, microsomes from insect cells containing human CYP1A2 (■) or CYP3A4 (♦) at a final concentration of 50 and 30 nM, respectively, were incubated with clopidogrel as described under Experimental Procedures. At the indicated times, aliquots were removed, and the remaining clopidogrel was quantified as described previously. The data are presented as the mean ± S.D of three separate experiments. B, microsomes from human lymphoblast cells containing either CYP3A4 (♦) or CYP1A2 (■) at a final concentration of 90 or 110 nM, respectively, were incubated with clopidogrel as described above. The data shown are the mean ± S.D. of three experiments.
Stimulation of the Rate of CYP3A4 Clopidogrel Oxidation by Cytochrome b5.
Cyt b5 can stimulate the oxidation of selected substrates by P450 in a reconstituted purified system when purified cyt b5 is incorporated into liver microsomes (Peterson and Prough, 1986; Gruenke et al., 1995;Vergeres et al., 1995). To determine whether purified amphipathic cytb5 could stimulate the CYP3A4 catalyzed oxidation of clopidogrel in insect cell microsomes, which contained CYP3A4/P450 reductase in a molar ratio of 1:0.15, pure cytb5 was incorporated into insect cell microsomes. Cyt b5 was not detectable in the microsomes used in these studies. Exogenously added cytb5 maximally increased the rate of clopidogrel oxidation from 7 to 11 nmol min−1nmol P450−1 at a final molar ratio of CYP3A4/cytb5 of 1:0.25. Higher concentrations of cyt b5 up to a CYP3A4/cytb5 molar ratio of 1:2 did not further increase the oxidation of clopidogrel. These results demonstrate that, as has been reported for many other substrates, cytb5 is able to stimulate the metabolism of clopidogrel by CYP3A4.
Determination of the Ks between Human CYP3A4 and Clopidogrel.
CYP3A4 metabolizes 30 to 40% of drugs and steroids in the liver, and multiple substrates often compete for available CYP3A4 (Guengerich, 1995; Thummel and Wilkinson, 1998). To improve the prediction of clopidogrel interaction with other CYP3A4 substrates, the dissociation constant between ferric CYP3A4 and clopidogrel was measured. Addition of clopidogrel to ferric CYP3A4 caused a change in spin state from a low spin form in which water is the 6th ligand to a high spin form in which water is no longer present and the heme is pentacoordinate. This is manifested by a shift in the Soret peak of the P450 spectrum resulting in an increase in absorbance at 385 nm and a decrease at 420 nm (Type I spectral change). The amplitude of the absorbance change between 385 and 420 nm increased hyperbolically as clopidogrel was added, giving a total absorbance change of 0.008 after the addition of 45 μM clopidogrel to 0.1 μM ferric CYP3A4 (Fig.3). The data were fitted to a reciprocal plot (Fig. 3, insert), and the Ksbetween clopidogrel and oxidized human CYP3A4 in insect cell microsomes was calculated as 12 μM.
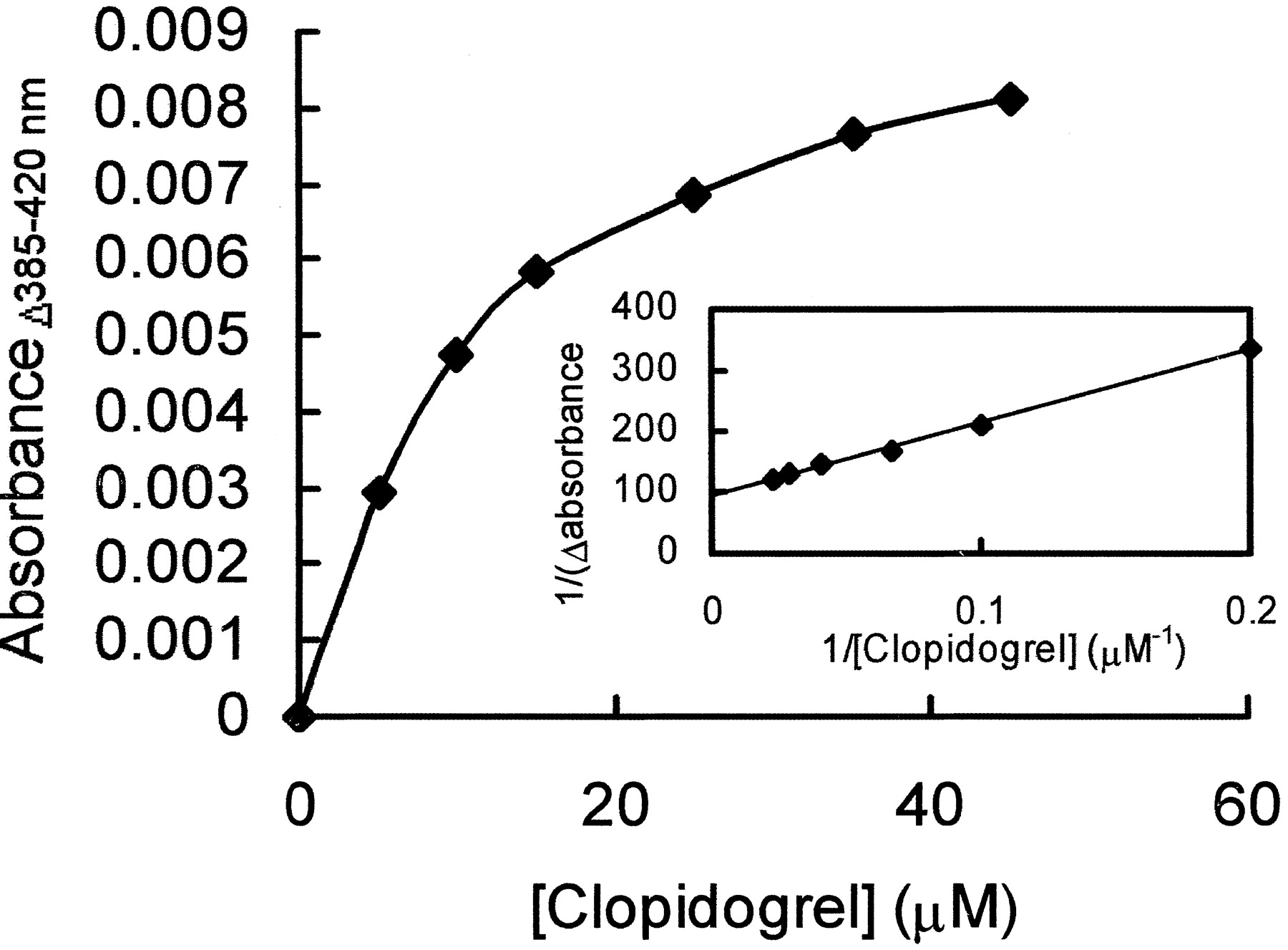
Change in absorbance of CYP3A4 on addition of clopidogrel.
Ferric CYP3A4 (1 μM) was titrated with increasing amounts of clopidogrel, and the resulting change in absorbance between 385 and 420 nM was recorded. The data shown are the mean ± S.D. of three experiments. Insert, reciprocal plot of the measured change in absorbance. A maximal absorbance change (Amax) of 0.011 was determined from the intercept, and the slope of the line was equal toKs/Amax
Inhibition of the CYP3A4 and 3A5 Catalyzed Metabolism of Clopidogrel by the Acid and Lactone Forms of Atorvastatin.
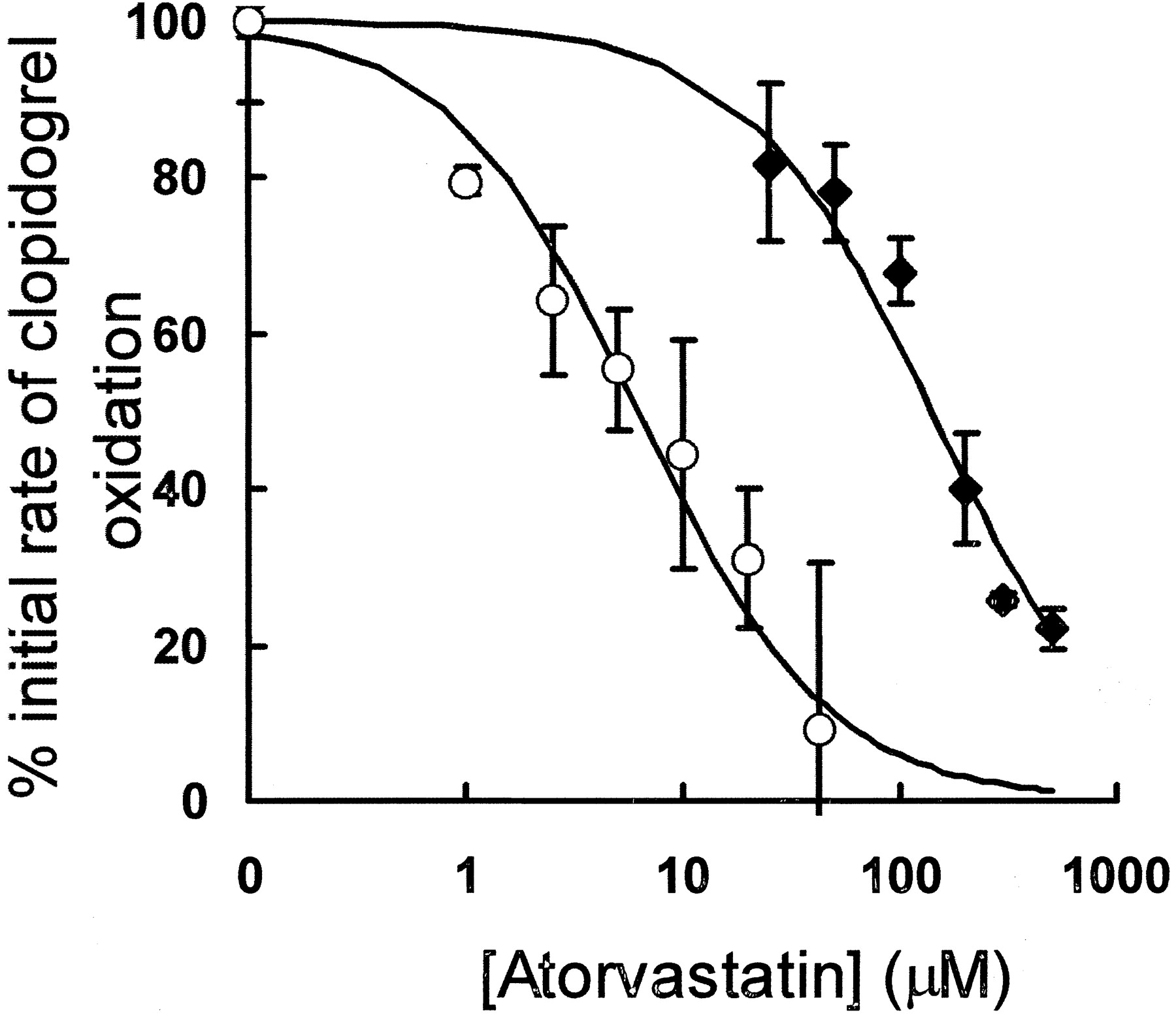
Since clopidogrel and atorvastatin are often administered simultaneously, it was of interest to determine whether atorvastatin acid or atorvastatin lactone could inhibit clopidogrel metabolism in vitro. The rate of clopidogrel oxidation by CYP3A4 in human lymphoblastoid microsomes was measured in the presence of different atorvastatin concentrations. As the concentration of the acid and lactone forms of atorvastatin increased, the rate of clopidogrel oxidation decreased (Fig. 4).

Inhibition of the rate of CYP3A4-dependent clopidogrel oxidation by atorvastatin acid and atorvastatin lactone.
CYP3A4 expressed in human B lymphoblasts (45 nM) was incubated with clopidogrel (50 μM) and increasing amounts of either atorvastatin acid (♦) or atorvastatin lactone (○) as described underExperimental Procedures. The data shown for both acid and lactone forms are from two separate experiments, and the solid lines are fits to the data.
Atorvastatin acid inhibited 80% of clopidogrel oxidation at a 10-fold molar excess. In contrast, atorvastatin lactone inhibited 90% CYP3A4-dependent clopidogrel oxidation at an equimolar concentration. The inhibition caused by atorvastatin lactone and acid could be fit to a hyperbolic curve that gave inhibition constants (Ki) of 6 and 140 μM for atorvastatin lactone and acid, respectively. These values are consistent with the known Michaelis-Menten constants of 1.4 and 25 μM for atorvastatin lactone and acid hydroxylation, respectively, in human liver microsomes and indicate that the lactone is the physiological substrate of CYP3A4 (Jacobsen et al., 2000).
The effect of the acid and lactone forms on the metabolism of clopidogrel by CYP3A5 was also investigated. Incubation of CYP3A5 and clopidogrel with different concentrations of atorvastatin acid or lactone showed a decrease in activity as the concentration of atorvastatin increased. The pattern of inhibition was similar to the inhibition of CYP3A4 described above and the data for CYP3A5 could be fit to give inhibition constants of 12 and 185 μM for atorvastatin lactone and atorvastatin acid, respectively.
In this reaction clopidogrel and atorvastatin are two alternate substrates competing for the same active site. Therefore one compound can be considered as a substrate, and the second substrate can be treated as a classical competitive inhibitor (Cha, 1968). Since theKs between clopidogrel and CYP3A4 had been experimentally determined and theKm of atorvastatin was known from the literature, it was of interest to calculate theKm of clopidogrel and compare it to the experimentally determined Ks(Jacobsen et al., 2000).
The following equation relates the experimentally determined rate of clopidogrel oxidation to its Km andVmax and theKm andVmax of atorvastatin (Cornish-Bowden, 1979):
The data shown in Fig. 4 were fitted to eq. 1 for both the acid and lactone forms using the published values for theKm of atorvastatin acid and lactone and the solver function in Microsoft Excel (Jacobsen et al., 2000). The calculated Km andVmax for clopidogrel are 14 ± 1 μM and 6.7 ± 1 nmol of clopidogrel oxidized min−1 nmol P450−1, in good agreement with the experimentally determined values for theKs (12 μM) and rate of clopidogrel metabolism by P450 expressed in human lymphoblast cells and dexamethasone-induced rat liver microsomes (4 nmol min−1 nmol CYP3A4−1).
Discussion
Our in vitro results demonstrate that CYP3A4 and 3A5 oxidize clopidogrel much faster than any other human P450. These findings support the clinical studies, which suggest that clopidogrel is metabolized in humans by CYP3A4, since the antiplatelet activity of clopidogrel can be inhibited by the CYP3A4 substrates erythromycin, troleadomycin, and atorvastatin and enhanced by the CYP3A4 inducer rifampin (Lau et al., 2002). In addition, clopidogrel metabolism is inhibited by an equimolar ratio of atorvastatin lactone that acts as a competing alternate substrate at the putative substrate binding site of CYP3A4. Since cerivastatin, lovastatin, and simvastatin bind to CYP3A4 with an affinity similar to that of atorvastatin and are present in vivo at similar concentrations to atorvastatin, it is anticipated that these HMG-CoA reductase inhibitors will also diminish the activation of clopidogrel (Igel et al., 2001).
Identification of CYP3A4 and 3A5 as the major human cytochrome P450 isozymes that metabolize clopidogrel and delineation of the dissociation constant between clopidogrel and these cytochrome P450 isozymes establishes a theoretical basis for predicting drug interactions between clopidogrel and other CYP3A4 substrates. The extent of the competitive inhibition of one substrate by another is a function of the relative in vivo intrahepatic concentration of the two substrates and their relative affinity for the substrate binding site of the cytochrome P450. A tabulation of the spectral dissociation constants and inhibitory constants between human CYP3As and numerous drugs has been compiled (Thummel and Wilkinson, 1998). The dissociation constants vary from 0.25 nM for clotrimazole to 83 μM for rapamycin. Clopidogrel with an intermediate binding affinity for CYP3A4 (Ks = 10 μM) would, therefore, be likely to have its metabolism inhibited by tight-binding substrates such as the azole antifungals, selected immunosuppressants, and human immunodeficiency virus protease inhibitors, which also have significant in vivo levels. In contrast, clopidogrel, which exists at low concentrations in vivo because of its rapid hydrolysis by esterases to the inactive carboxylic acid, would only be expected to inhibit other substrates, which also bind with modest to poor affinity and are present in low concentrations in vivo (Reist et al., 2000; product information from Sanofi-Synthelabo, Montpellier, France). If the in vivo concentration of a given drug is available or can be estimated and its dissociation constant with CYP3A4 is known, these values can be compared with the analogous values for clopidogrel, and the likelihood that a drug will modify the metabolism of clopidogrel can be estimated. In view of the great variability in human drug metabolism, this would be a semiquantitative prediction. In addition, compounds that induce CYP3A, such as rifampin and “St. John's Wort,” would promote the antiplatelet effects of clopidogrel whereas the CYP3A inhibitors such as the macrolide antibiotics, erythromycin, and troleandomycin, would diminish the activity of clopidogrel (Lau et al., 2002).
Attempts to observe clopidogrel metabolites using rat liver microsomes were unsuccessful so clopidogrel disappearance was used to measure P450-dependent clopidogrel metabolism. A previous study that measured clopidogrel metabolism by the loss of the C14-labeled clopidogrel identified P450 1A in rat liver microsomes as the main oxidizer of clopidogrel (Savi et al., 1994). In contrast, our studies suggested rat CYP3A, not 1A2, was responsible for clopidogrel oxidation. As a result, most experiments were designed to compare the rate of clopidogrel metabolism between rat CYP3A and CYP1A2. In three different microsomal systems (rat liver, insect cell, human lymphoblast), CYP3A4 was observed to metabolize clopidogrel more rapidly than CYP1A2. We cannot explain the difference between our results and the previously published studies, but they are consistent with clinical studies that show clopidogrel metabolism is inhibited by the CYP3A4 inhibitors, erythromycin and troleandomycin, and stimulated by the CYP3A4-inducer rifampin (Lau et al., 2002).
Although CYP1A2 in insect cell microsomes metabolized clopidogrel at a very low rate, virtually no clopidogrel was oxidized by CYP1A2 in human B lymphoblasts or microsomes prepared from rats treated with β-napthoflavone. This suggests that, at physiological molar ratios of CYP1A2/P450 reductase, CYP1A2 does not metabolize clopidogrel. Clopidogrel was also metabolized at low rates in insect cell microsomes containing CYP2B6. However, CYP2B6 typically accounts for <1% of the total hepatic P450 in the liver, so it is unlikely to play a major role in metabolizing clopidogrel because, in the majority of human livers, CYP3A4 and 3A5 comprise approximately 30 to 50% of the total hepatic P450. However, it is possible that clopidogrel may be metabolized to a considerable extent by CYP2B6 in patients with low hepatic concentrations of CYP3A4 and CYP3A5 and high levels of CYP2B6.
Studies by Crespi and Miller (1999) and Shaw et al. (1997) have shown that the activity of P450 in insect cell microsomes can be over 7-fold higher than the P450 activity for other systems, such as human liver microsomes, human B lymphoblast microsomes, or purified reconstituted systems. This is attributed to the high but variable molar ratio of P450/P450 reductase (1:1–5) in insect cell microsomes. In human and rat hepatic microsomes, the molar ratio is 20 P450:1 P450 reductase, and the transfer of electrons to P450 from reductase is considered to be rate limiting (Peterson and Prough, 1986). Human lymphoblast microsomes have a P450/P450 reductase molar ratio of 1:0.05–0.2, which is closer to the physiological value, and these microsomes metabolize clopidogrel at a rate similar to that of hepatic microsomes from dexamethasone-treated rats.
Acknowledgments
We thank Drs. P. Hollenberg and U. M. Kent for the gift of rat liver microsomes and assistance in measuring the metabolism of testosterone, erythromycin, and ethoxyresorufin. Atorvastatin calcium was a generous gift from Pfizer. We would also like to gratefully acknowledge that our in vitro studies were prompted by Dr. Wei Lau's suggestion that clopidogrel might be activated by human CYP3A4.
Footnotes
-
Grant support: National Institutes of Health GM 35533 (L.W.), Veterans Administration Merit Review (L.W.)
- Abbreviations used are::
- P450
- cytochrome P450
- HMG-CoA
- 3-hydroxyl-3-methylglutaryl-coenzyme A
- HPLC
- high-pressure liquid chromatography
- cyt b5
- cytochrome b5
- Ks
- spectral dissociation constant
- CS-747
- 2-acetoxy-5-(α-cyclopropylcarbonyl-2-fluorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine
- Received July 22, 2002.
- Accepted September 30, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}