


Abstract
Drug-induced liver injury (DILI) is of concern to the pharmaceutical industry, and reliable preclinical screens are required. Previously, we established an in vitro bile acid–dependent hepatotoxicity assay that mimics cholestatic DILI in vivo. Here, we confirmed that this assay can predict cholestatic DILI in clinical situations by comparing in vitro cytotoxicity data with in vivo risk. For 38 drugs, the frequencies of abnormal increases in serum alkaline phosphatase (ALP), transaminases, gamma glutamyltranspeptidase (γGT), and bilirubin were collected from interview forms. Drugs with frequencies of serum marker increases higher than 1% were classified as high DILI risk compounds. In vitro cytotoxicity was assessed by monitoring lactate dehydrogenase release from rat and human sandwich-cultured hepatocytes (SCRHs and SCHHs) incubated with the test drugs (50 μM) for 24 hours in the absence or presence of a bile acids mixture. Receiver operating characteristic analyses gave optimal cutoff toxicity values of 19.5% and 9.2% for ALP and transaminases in SCRHs, respectively. Using this cutoff, high- and low-risk drugs were separated with 65.4–78.6% sensitivity and 66.7–79.2% specificity. Good separation was also achieved using SCHHs. In conclusion, cholestatic DILI risk can be successfully predicted using a sandwich-cultured hepatocyte–based assay.
Introduction
Drug-induced liver injury (DILI) is a potentially serious adverse reaction that leads to the dropout of candidate compounds from drug development processes and the withdrawal of pharmaceuticals from clinical use (Kaplowitz, 2013). DILI can severely damage the liver, resulting in liver transplantation in worst case scenarios; hence, it is essential to identify, remove, and/or assign alerts for possible DILI risk compounds at all stages of the drug development process.
The accumulation of bile acids (BAs) within hepatocytes has been suggested as an underlying mechanism of cholestatic DILI (Stieger et al., 2000; Fattinger et al., 2001; Byrne et al., 2002; Kostrubsky et al., 2006). The bile salt export pump (human BSEP/rat Bsep), which is localized on the apical side of the hepatocyte plasma membrane, plays a major role in the excretion of BAs from the liver to the bile (Meier and Stieger, 2002). Several genetic mutations of BSEP are associated with progressive familial intrahepatic cholestasis type 2, and cause severe intracellular accumulation of BAs within the liver (Strautnieks et al., 1998). A similar phenotype also occurs if BSEP function is inhibited or its expression is suppressed by drugs, leading to cholestatic or mixed-type DILI (Roman et al., 2003; Dawson et al., 2012). This concept is widely accepted; however, it is still difficult to detect cholestatic DILI risk using preclinical animals. Notably, Bsep knockout mice develop severe cholestasis when fed a BA-enriched diet, but display only mild cholestasis when fed a normal diet. This outcome is explained by the fact that 1) endogenous BAs are less toxic to rodents than humans because taurine-conjugated BA species (nontoxic) were predominant in rodents, whereas glycine-conjugated BAs (toxic) were predominant in humans (Thomas et al., 2008; Marion et al., 2012), and 2) adaptive changes in the expression levels of enzymes and transporters occur in Bsep knockout mice (Wang et al., 2003, 2009).
A membrane vesicle assay is considered as a simple and suitable alternative method of predicting the cholestatic DILI potential of test drugs. Previous studies have demonstrated that many drugs that cause cholestatic DILI are potent inhibitors of BSEP (Morgan et al., 2010; Dawson et al., 2012; Warner et al., 2012; Pedersen et al., 2013). Nonetheless, it was gradually indicated that the BSEP-based vesicle assay might misestimate the risk of cholestatic DILI, likely because cell-free systems lack drug metabolism pathways and other BA efflux transporters [such as multidrug resistance-associated protein 3 (MRP3) and MRP4] (Dawson et al., 2012).
Historically, predictions of cholestatic DILI risk have depended mainly on membrane vesicle assays, as described earlier (Morgan et al., 2010, 2013; Dawson et al., 2012; Pedersen et al., 2013; Kock et al., 2014). Although this method is easy to perform and suitable for high-throughput screening, it does have some limitations, as discussed elsewhere (Morgan et al., 2013; Pedersen et al., 2013; Kock et al., 2014). It was also claimed that sandwich-cultured hepatocyte (SCH)–based transport assays are more suitable than membrane vesicle assays to precisely differentiate BSEP inhibitors that result in relatively mild DILI from those associated with more severe DILI (Pedersen et al., 2013).
To overcome the less-toxic nature of BA species in preclinical animals and the shortcomings of the membrane vesicle assay described earlier, we established a unique cell-based toxicity assay using SCHs in combination with titrated human BA species (Ogimura et al., 2011). The rationale of using SCHs is that drug-metabolizing enzymes and transporters are well maintained compared with standard culture conditions (Swift et al., 2010). If a test drug and/or its metabolite inhibits BA efflux from SCHs, the accumulation of BAs eventually induces cell death and is detected by the release of lactate dehydrogenase (LDH) into the medium. Indeed, in a previous study, we have demonstrated that 11 of the 26 test drugs examined exhibited significant toxicity in rat SCHs (SCRHs) only in the presence of such human BA compositions (Ogimura et al., 2011). Although most (8 of 11) of these toxic drugs were known BSEP/Bsep inhibitors, it is not yet known if our in vitro assay is capable of predicting cholestatic DILI risk in clinical situations; therefore, the aim of this study was to answer this question.
First, we performed an exhaustive search of the Japanese Adverse Drug Event Report database to refine test drug candidates, and then surveyed documents of interest to obtain the actual frequencies of serum marker increases. Finally, these data were compared with the in vitro toxicity data obtained using SCRHs and human SCHs (SCHHs).
Materials and Methods
Selection of Test Drugs.
To select test drugs, we surveyed the Japanese Adverse Drug Event Report database, which is operated by Japan's Pharmaceuticals and Medical Devices Agency (PMDA). From a total of 1,866,993 cases (reported between August 2004 and August 2013), 421,904 were extracted as drug-related adverse events. From these cases, 1984 drugs were identified as the most likely candidates, according to the guideline issued by the PMDA. The drugs of interest were scored according to the number of cases using the following keywords: hepatocellular injury [“liver injury,” “liver dysfunction,” “serum albumin concentration,” “total protein in serum,” “aspartate transaminase (AST),” “alanine transaminase (ALT),” “transaminase,” “nonalcoholic steatohepatitis,” “hepatic cirrhosis,” “liver inflammation,” “steatosis,” “acute or chronic liver failure,” “hepatic fibrosis,” “liver carcinoma,” and “fulminant hepatic failure”] and cholestasis [“jaundice,” “hyperbilirubinemia,” “alkaline phosphatase (ALP),” and “gamma glutamyltranspeptidase (γGT)].” The 243 drugs were then arranged in descending order of the rate of cholestatic DILI, which was calculated as follows: rate of cholestatic DILI = (cases with cholestasis keywords) / [(cases with cholestasis keywords) + (cases with hepatocellular injury keywords)]. Finally, 38 drugs that covered a broad range of cholestatic DILI incidences were selected from the list. As for the test set in SCHHs, six drugs which showed strong hepatocyte toxicity independent of BAs in SCRHs were first excluded from 38 drugs, and then a representative 22 drugs (12 drugs which showed BA-dependent hepatocyte toxicity and 10 drugs which did not show any toxicity; in SCRHs) were selected.
Frequency of Serum Test Abnormalities.
To calculate the frequencies of serum marker increases, we collected interview forms for 38 brand-name drugs from pharmaceutical companies. Abnormal serum levels of ALP, transaminases (AST/ALT), γGT, and bilirubin were selected as markers of a particular type of DILI. The incidences of these serum biomarkers and the numbers of patients (including infants) examined were extracted from the interview forms. The clinical studies based on: 1) combination therapy (e.g., antibiotics with proton pump inhibitors) and 2) non-Japanese patients were excluded from the number of cases. The frequencies of increases in the markers were calculated as follows: frequency of ALP, transaminases, γGT, or bilirubin abnormalities (percentage) = (number of cases with the serum test abnormality) / (number of patients enrolled in the clinical studies in Japan) × 100.
Materials.
BAs and test compounds were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan), Sigma-Aldrich (St. Louis, MO), or Calbiochem (Darmstadt, Germany). Williams’ medium E (WME), antibiotic-antimycotic solution, and GlutaMAX were purchased from Invitrogen (Carlsbad, CA). Insulin was purchased from Sigma-Aldrich. Matrigel, insulin, human transferrin, and selenous acid premix culture supplement were purchased from BD Biosciences (San Jose, CA). Collagenase and dexamethasone were purchased from Wako Pure Chemical Industries Ltd. All other chemicals and solvents were of analytical grade, unless otherwise noted.
Animals.
Male Sprague-Dawley rats (SLC Japan Inc., Tokyo, Japan), aged 7–8 weeks, were used throughout the study. The animals were treated humanely in accordance with the guidelines issued by the National Institutes of Health (Bethesda, MD). In addition, all procedures were approved by the Animal Care Committee of Chiba University (Chiba, Japan).
Rat and Human Sandwich-Cultured Hepatocytes.
Tissue culture (96-well) plates were precoated with type 1 collagen (BD Biosciences) for at least 1 hour prior to the preparation of hepatocyte cultures. Rat hepatocytes were isolated using a two-step perfusion method, as described previously by our group (Ogimura et al., 2011; Susukida et al., 2015). Isolated hepatocytes were seeded onto collagen-coated (1.5 mg/ml, pH 7.4) 96-well plates at a density of 0.48 × 105 cells/well in “plating medium” consisting of WME containing 5% fetal bovine serum, 0.1 µM dexamethasone, 4 mg/l insulin, 2 mM GlutaMAX, 15 mM HEPES (pH 7.4), penicillin (100 units/ml), and streptomycin (100 µg/ml). At 1.5 hours after seeding, the medium was aspirated, and fresh “plating medium” was added to each well. In brief, 24 hours after plating, hepatocytes were overlaid with Matrigel (0.25 mg/ml) dissolved in ice-cold “culture medium” consisting of WME containing 1% insulin, human transferrin, and selenous acid, 0.1 μM dexamethasone, 2 mM GlutaMAX, penicillin (100 units/ml), and streptomycin (100 µg/ml). Thereafter, the medium (WME) was changed daily until 4 days after cell seeding. For SCHHs, cryopreserved human hepatocytes (Life Technologies, Grand Island, NY) were thawed and seeded according to the manufacturer's protocol. Thawed hepatocytes were poured into CHRM Medium (Life Technologies) at 37°C. The cells were centrifuged at 100g for 10 minutes at room temperature and resuspended in “plating medium” consisting of WME containing 5% fetal bovine serum, 0.1 µM dexamethasone, 4 mg/l insulin, 2 mM GlutaMAX, 15 mM HEPES (pH 7.4), penicillin (100 units/ml), and streptomycin (100 µg/ml). Then, hepatocytes were seeded onto a collagen-coated 96-well plate at a density of 0.48 × 105 cells/well. At 4 hours after seeding, the medium was aspirated, and hepatocytes were overlaid with Matrigel (0.25 mg/ml) dissolved in ice-cold “culture medium”. The medium (WME) was changed daily until 5 days after cell seeding. Both SCRHs and SCHHs were maintained at 37°C in a humidified atmosphere of 95% air/5% CO2. Information of used lots of human hepatocytes is described in Table 1.
Donor information of used lots of human hepatocytes
Cytotoxicity Assay.
SCRHs and SCHHs were exposed to each test compound in the presence or absence of a BA mixture comprising the 12 BAs shown in Table 2. After exposure to the test compounds for 24 hours, cytotoxicity was assessed by measuring the activity of LDH released from damaged cells (LDH sample) using the LDH-Cytotoxic Test (Takara Bio Inc., Shiga, Japan). LDH activity was expressed as a percentage of maximum LDH activity in 0.25% Triton X-100–solubilized lysates of control SCHs (LDH Triton X-100), using the following equation: cell toxicity (%) = (LDH sample − LDH blank) / (LDH Triton X-100 − LDH blank) × 100. The LDH blank value was determined using untreated SCHs.
Composition of the 1 × BA mixture standard
Concentrations of each BA are set based on the standard BA constituents of human serum (Scherer et al., 2009).
Receiver Operating Characteristic Analysis.
Statistical analyses of the predictabilities of the drug-induced frequencies of ALP, transaminases, γGT, and bilirubin increases in human serum were performed using receiver operating characteristic (ROC) curves in GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA). The ROC curves were generated by computing the paired true-positive and false-positive rates for all possible thresholds of the cell toxicity assays using SCRHs and SCHHs. In ROC curves, the predictions that occur toward the top-left corner are indicative of high true-positive and low false-positive rates. This approach generates a threshold where the false-positive rate is zero and the true-positive rate is a fraction between 0% and 100%.
Results
Investigation of the Clinical Risk of Cholestatic DILI Compounds.
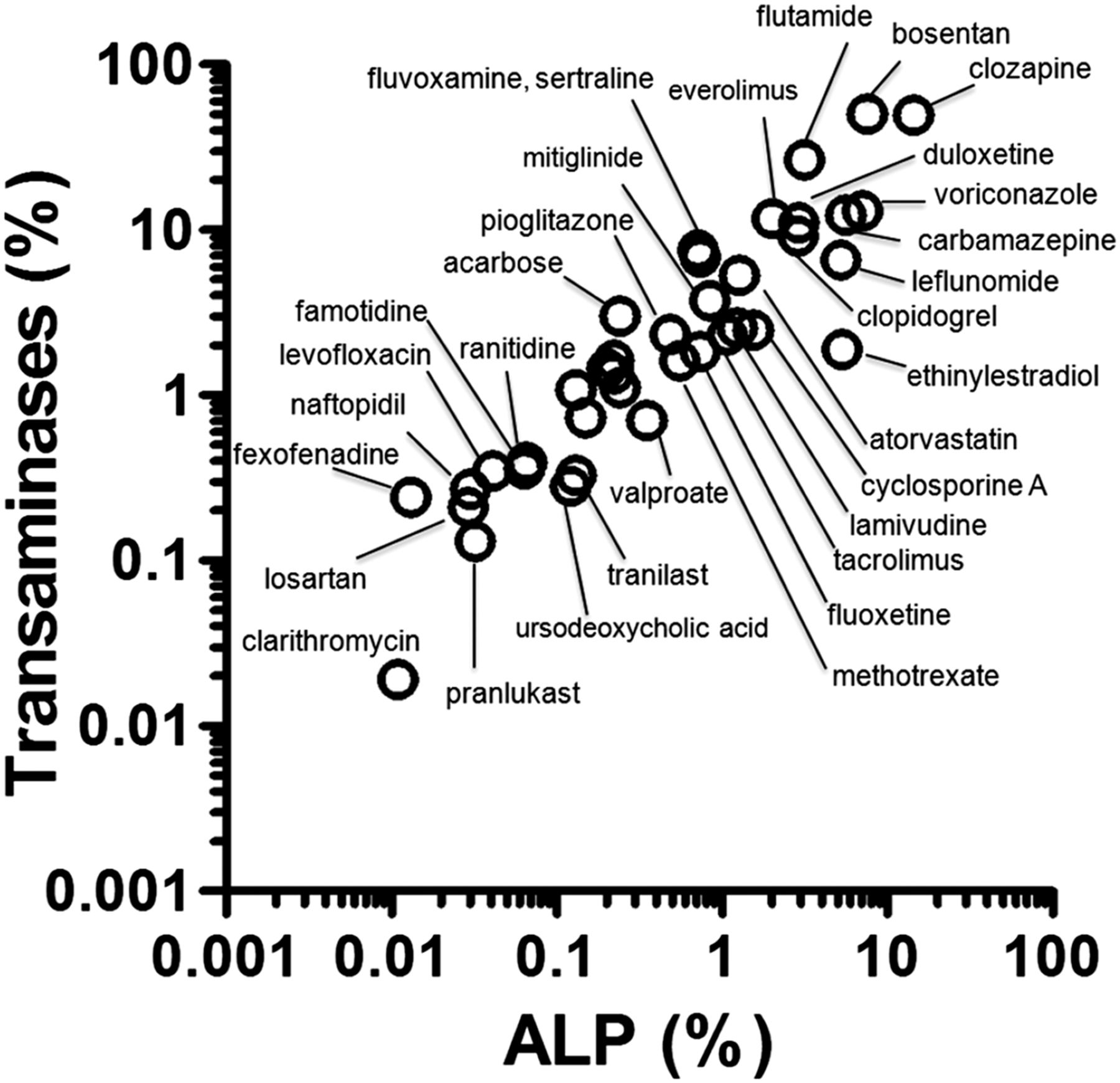
To identify the test drugs, we surveyed the PMDA’s voluntary adverse reaction database. From the top 243 drugs related to DILI, 38 were arbitrarily selected to cover the whole range, and a detailed survey of the clinical frequencies of cholestatic DILI was performed using the information in interview forms (Table 3). The frequency of ALP increases ranged from 0.011% (clarithromycin) to 14.3% (clozapine), with a median of 0.52%. The frequency of transaminase (ALT/AST) increases ranged from 0.019% (clarithromycin) to 50.0% (bosentan), with a median of 1.85%. The frequency of γGT increases ranged from 0.0040% (tranilast) to 18.1% (carbamazepine), with a median of 0.61%. The frequency of bilirubin increases ranged from 0.0040% (clarithromycin) to 2.5% (bosentan), with a median of 0.19%. Notably, the frequencies of ALP and transaminases [these serum markers are used in the diagnosis and management of DILI (Chalasani et al., 2014)] were positively correlated [linear regression after logarithmic conversions; r2 = 0.840 for ALP vs. transaminases (Fig. 1)]. The other relationships between frequencies of clinical markers (e.g., ALP vs. γGT) are shown in Supplemental Fig. 1.
List of the drugs used in the study and their in vivo profiles and in vitro toxicities obtained in SCH assays

Relationship between the frequencies of serum marker increases in patients. Frequencies of increases in the serum levels of ALP and transaminases. The frequencies were calculated for 38 selected drugs based on the numbers of cases reported in the interview forms. Frequency data are shown in Table 3.
Optimization of the BA Mixture Concentration.
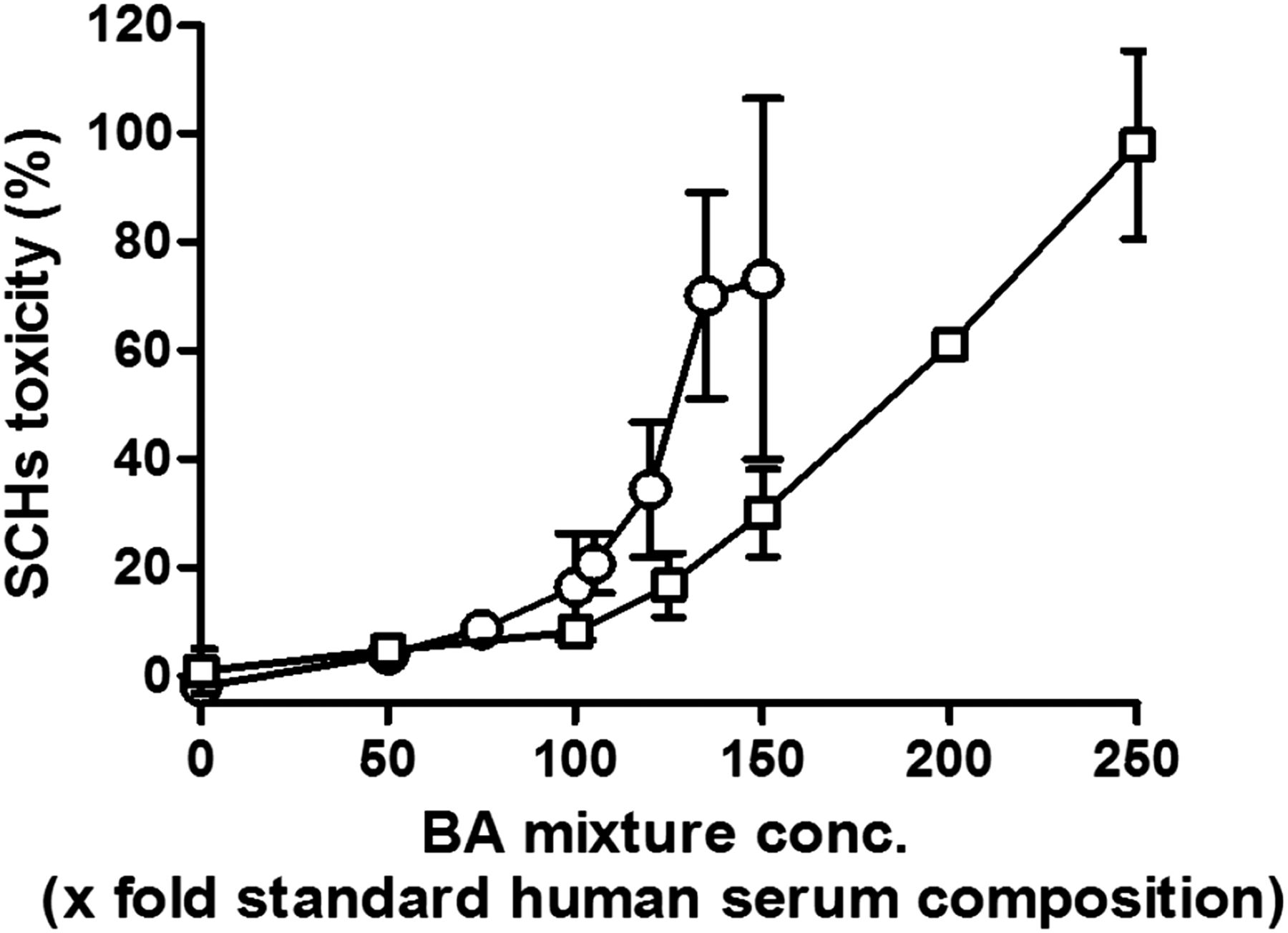
Before performing BA-dependent drug toxicity assays, we examined the sensitivities of SCRHs and SCHHs to various concentrations of the standard (1×) BA mixture of human serum (Scherer et al., 2009) (Table 2). In both cell types, toxicity began to rise at 100× concentrations, and increased in a concentration-dependent manner. In our assay condition, SCRHs was more susceptible compared with SCHHs (Fig. 2). To detect subtle changes in BA accumulation–dependent enhancement of the toxicity, we used a 115× BA mixture for SCRHs and a 150× BA mixture for SCHHs in subsequent assays, where the basal toxicity of 20–30% was expected. These optimized exogenous BAs to SCRHs and SCHHs in this study were significant excess than in clinical situations (at least 115× human serum concentrations). This rational was supported by the downregulation of mRNA expression levels of BA uptake transporters (i.e., Ntcp and Oatp1a1) in SCRHs (Susukida et al., 2015).

BA concentration–dependent toxicity in SCRHs and SCHHs. SCRHs (open circles) and SCHHs (open squares) were cultured for 4 days (rat) or 5 days (human), then treated with various concentrations (conc.) of the BA mixture (0–150× standard BA mixture for SCRHs and 0–250× standard BA mixture for SCHHs) for 24 hours. The composition and concentration of the standard (1×) BA mixture is shown in Table 2. Cell toxicity was determined by measuring the activity of LDH released into the medium. Data are represented as the mean ± S.E.M. of n = 3.
Evaluation of Drug Toxicity in SCRHs and SCHHs.
The toxicities of the 38 drugs to SCRHs were determined using drug concentrations of 50 μM (except cyclosporine A used as positive control, 10 µM) and the 115× BA mixture. With the exceptions of seven drugs (amiodarone, clozapine, duloxetine, ethinyl estradiol, everolimus, fluoxetine, and sertraline), the drugs showed minimum toxicity to SCRHs in the absence of the BA mixture (Fig. 3A). The toxicities of the drugs were more or less enhanced in the presence of the 115× BA mixture (Fig. 3A). Similarly, the toxicities of the arbitrarily selected 22 drugs to SCHHs were determined using drug concentrations of 50 μM (except cyclosporine A used as positive control, 10 µM) and the 150× BA mixture. With the exception of two drugs (amiodarone and tacrolimus, in Hu4197), the drugs showed minimum toxicity to SCHHs in the absence of the BA mixture (Fig. 3, B–D). The toxicities of the drugs to SCHHs were more or less enhanced in the presence of the 150× BA mixture (Fig. 3, B–D). When correlation analyses of the toxicities to each lot of human hepatocytes in the absence and presence of the BA mixture of the 22 tested drugs were performed, a positive correlation was observed in the presence of BAs (Supplemental Fig. 2, D–F) but not in the absence of BAs (Supplemental Fig. 2, A–C). However, some drugs (clopidogrel, leflunomide, and ticlopidine) showed lot variation for BA-dependent hepatocyte toxicity in SCHHs.

In vitro toxicities of the selected drugs to SCRHs and SCHHs in the absence or presence of the BA mixture. The SCRHs were cultured for 4 days (A) and SCHHs were cultured for 5 days [(B) Hu1437; (C) Hu1524; (D) Hu4197]. They were treated with cyclosporine A (10 µM) or the other test drugs (50 μM) in the absence (open bars) or presence (closed bars) of the BA mixture (115× for SCRHs and 150× for SCHHs). The basal toxicities in the absence and presence of the BA mixture were obtained and subtracted from the corresponding data obtained in the presence of the test drugs. Data are represented as the mean ± S.E.M. of n = 3 for SCRHs and n = 2–3 for SCHHs.
Correlation of Rat and Human Sandwich Hepatocyte Toxicity Data.
To determine if the SCRHs and SCHHs responded similarly to the drugs, correlation analyses of the toxicities to each cell type in the absence and presence of the BA mixture were performed for the 22 drugs tested in common (Fig. 4). Although the correlation between the rat and human cells was poor in the absence of the BA mixture (Fig. 4, A–C), a positive correlation was observed in the presence of this mixture (Fig. 4D: y = 0.4492x – 2.142, r2 = 0.7516; Fig. 4E: y = 0.6525x + 4.501, r2 = 0.7543; Fig. 4F: y = 0.6242x + 3.698, r2 = 0.7431).

Correlation between the in vitro BA-dependent hepatocyte toxicities of the selected drugs to SCRHs and SCHHs. The in vitro toxicities of the common 22 drugs to SCRHs and SCHHs in the absence (A–C) or presence (D–F) of the BA mixture. The inset of each graph (D–F) shows drugs sitting on the x- and y-axes in the enlarged scale (within 0–15% of cell toxicity; drug numbers correspond to those shown in Table 3). The original data are shown in Fig. 3. A positive correlation [y = 0.4492x – 2.142, r2 = 0.7516 (D); y = 0.6525x + 4.501, r2 = 0.7543 (E); y = 0.6242x + 3.698, r2 = 0.7431(F)] was observed in the presence of the BA mixture.
Prediction of Clinical Risk from the In Vitro Data.
To evaluate if the in vitro toxicity data reflected the clinical risk of cholestatic DILI, the relationships between the in vitro and in vivo data were examined. Most of the drugs with lower frequencies of clinical ALP and transaminase increases showed minimal BA-dependent drug toxicity to SCRHs, whereas those with higher frequencies of clinical ALP and transaminase increases showed enhanced BA-dependent hepatocyte toxicity (Fig. 5, A and E). Similar tendencies were also observed when the frequencies of the ALP and transaminase increases were plotted against the BA-dependent hepatocyte toxicities of the drugs to SCHHs (Figs. 5, B–D and F–H). To evaluate the usefulness of the in vitro assay further, ROC analysis was performed. The clinical increases in serum markers of 1.0% were used to separate the drugs into low- and high-risk groups. As a result, by setting an in vitro toxicity cutoff value of 19.5% for SCRHs, the drugs with a high risk of clinical ALP increases (>1.0%) were correctly sorted with 78.6% sensitivity and 79.2% specificity (Fig. 5A; Table 4). Similarly, by setting an in vitro toxicity cutoff value of 9.2% for SCRHs, the drugs with a high risk of clinical transaminase increases (>1.0%) were correctly sorted with 65.4% sensitivity and 66.7% specificity (Fig. 5E; Table 4). The study of SCRHs was replicated at least two times, and therefore the represented data were shown. The replicated trial in SCRHs successfully predicted the clinical frequencies of serum marker increases for most drugs, and therefore the reproducibility of the work has been established (data not shown). In SCHHs, approximately 70% sensitivity and 80% specificity for the prediction of both clinical ALP increases and transaminase (>1.0%) increases were obtained from the analyses of Hu1524 and Hu4197 toxicity data (Fig. 5,C, D, G, and H; Table 4). However, analysis of the other lot of SCHH (Hu1437) toxicity data resulted in poor sensitivity (less than 50% for both serum markers) because of the lot variation of clopidogrel-, leflunomide-, and ticlopidine-induced BA-dependent hepatocyte toxicity (Fig. 5, B and F; Table 4). The clinical frequency of γGT increase was also well predicted in both SCRHs and SCHHs (Supplemental Fig. 3, A–D; Supplemental Table 2). On the contrary, the frequency of serum bilirubin increase was not correlated with the in vitro toxicities in both SCRHs and SCHHs (Supplemental Fig. 3, E–H; Supplemental Table 2). Prediction of serum marker elevations was not possible in the absence of the BA mixture (Supplemental Fig. 4).
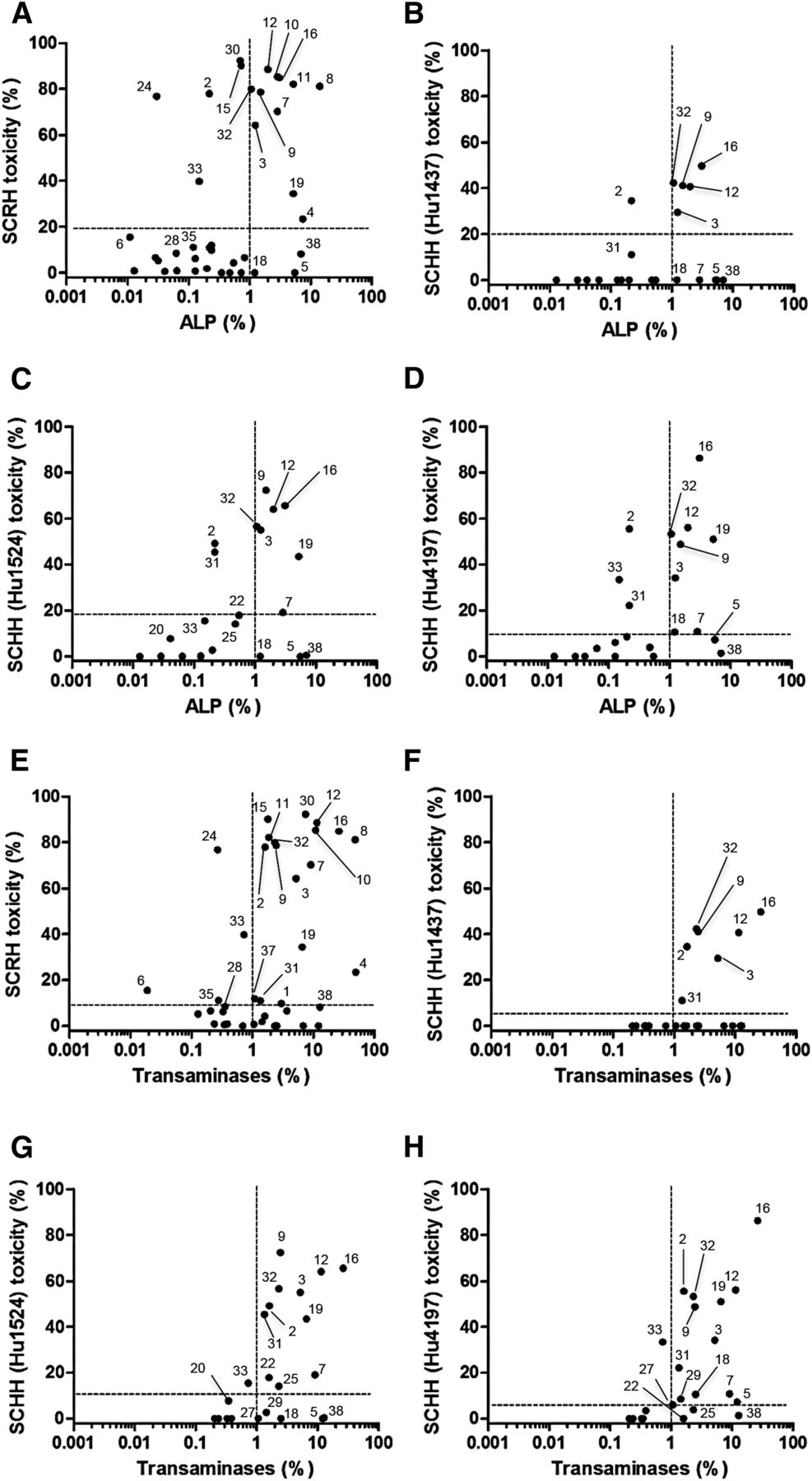
Relationship between the frequencies of serum marker increases in vivo and BA-dependent hepatocyte toxicity in vitro in the presence of BA mixture. The frequencies of increases in the serum levels of ALP (A–D) and transaminases (E–H) versus the in vitro toxicities to SCRHs (A and E) and SCHHs (B–D and F–H). The vertical dotted lines represent the borderline frequencies of 1%. The horizontal dotted lines represent the cutoff values determined by the ROC analysis, which gave the best separation of high- and low-risk drugs (see Table 4 for details). Drug numbers correspond to those shown in Table 3.
Predictabilities of the frequencies of in vivo serum test abnormalities from in vitro toxicity assays
Drugs (38 drugs for SCRHs and 22 drugs for SCHHs) were divided into two groups with lower or higher than 1.0% frequency of serum markers. An ROC analysis was performed to generate the best separation of the two groups using the in vitro drug toxicity data obtained in the presence of BA mixture. For example, by setting an in vitro toxicity cutoff value of 19.5%, drugs with a higher risk of ALP increase were correctly predicted with 78.6% accuracy, whereas drugs with a lower risk of ALP increase were correctly predicted with 79.2% accuracy.
To determine if the predictability of the in vitro assay was improved by considering the drug concentration in vivo, the maximum plasma concentrations (Cmax) and maximum unbound plasma concentrations (Cmax,u) were collected from the interview forms (Supplemental Table 1). These concentrations were multiplied by the in vitro toxicity data obtained in SCRHs and SCHHs and plotted against the in vivo frequencies of the increases in serum markers. However, such corrections did not improve the predictive capability of the in vitro assay (compare Supplemental Fig. 5, A and B with Fig. 5).
Discussion
We have previously established a unique SCH-based toxicity assay system focusing on cholestatic DILI (Ogimura et al., 2011) and now successfully confirmed here that the assay is particularly useful for the prediction of clinical serum marker abnormalities. The SCH-based toxicity assay described here has an advantage over the membrane vesicle transport assay in that it includes metabolic enzymes and BA efflux transporters other than BSEP/Bsep. For some drugs, their metabolites inhibit BSEP/Bsep more potently than the parent compound. For example, the IC50 of troglitazone against [3H]taurocholate ([3H]TC) uptake into isolated bile canalicular membrane vesicles from rat liver is 3.9 μM, whereas that of the sulfated conjugate is as low as 0.4–0.6 μM (Funk et al., 2001). Exposure of SCRHs to 10 μM troglitazone enhances BA-dependent hepatocyte toxicity significantly (Ogimura et al., 2011), which is likely explained by the extensive metabolism of the drug to its sulfate conjugate in these cells (Yang and Brouwer, 2014). Considering the importance of metabolic enzymes in our SCH-based toxicity assay, it sometimes gives distinct results because of the presence of large interspecies differences in drug-metabolizing enzyme expression and activity. Our group has reported that the parent form of glibenclamide (not used in the current study) might contribute to the BA-dependent hepatocyte toxicity in SCRHs (Ogimura et al., 2011), but it did not show BA-dependent hepatocyte toxicity in SCHHs (data not shown). That might be explained by the fact that glibenclamide was more extensively metabolized in human liver microsomes than in rat liver microsomes (Ravindran et al., 2013).
The BA-dependent hepatocyte toxicity of some drugs (clopidogrel, leflunomide, and ticlopidine) in Hu1437 was lower than that of the other two lots (Fig. 3B; Supplemental Fig. 2, D and E), resulting in poor sensitivity of the frequencies of in vivo serum test abnormalities (Fig. 5, B and F; Supplemental Fig. 3, B and F; Supplemental Table 2; Table 4). Given the parent form of these drugs showed minimum inhibitory effect against human BSEP (Morgan et al., 2013; see Supplemental Table 1), those metabolites possibly contribute to the toxicity and their intracellular amount might depend on the activity of metabolic enzymes in each lot. Notably, these three drugs are known to be metabolized by CYP2C19 and their metabolites themselves have pharmacologic effects (Bohanec Grabar et al., 2009; Farid et al., 2010; Nakkam et al., 2015), implying that CYP2C19 may be the candidate for producing toxic metabolites. According to the donor information of human hepatocytes (Table 1), the donor of Hu1437 was an elderly (70 years old) Caucasian female. A clinical investigation of the influences of age and gender on the disposition of the CYP2C19 substrate in Caucasians reported that elderly women (i.e., the donor of Hu1437) may have lower activity of CYP2C19 (Hooper and Qing, 1990; Kobayashi et al., 2004). Therefore, this could be one of many reasons for the absence of BA-dependent hepatocyte toxicity of these drugs (clopidogrel, leflunomide, and ticlopidine) in this lot. Interestingly, same tendency was also observed in SCRHs. These drugs did not show BA-dependent hepatocyte toxicity in another trial of SCRHs (Supplemental Fig. 6A). Although it is unknown which cytochrome P450 enzymes are involved in producing those toxic metabolites in rats, their amount may be influenced by the expression level of the involved cytochrome P450 enzyme in each trial of SCRHs.
The results of other drugs—except clopidogrel, leflunomide, and ticlopidine—presented here did not show both interspecies and lot variation, indicating that rat cells were a good alternate for human cells. This finding is partially attributable to the similar inhibition profiles of human BSEP and rat Bsep as reported previously (Morgan et al., 2010; Dawson et al., 2012). In a study of 56 compounds, it was found that the IC50 values of the majority of compounds toward [3H]TC uptake into membrane vesicles from Sf9 cells expressing human BSEP and rat Bsep were quite similar (Morgan et al., 2010). Moreover, in a study of 85 compounds, a close correlation (r2 = 0.94) was confirmed between the IC50 values of drugs toward [3H]TC uptake into membrane vesicles isolated from Sf21 cells expressing human BSEP and rat Bsep (Dawson et al., 2012).
One might expect that the prediction accuracy of an in vitro assay would be improved by considering the pharmacokinetic (PK) parameters of each drug. In a previous study, 95% of compounds with a steady-state plasma concentration/BSEP IC50 ratio ≥0.1 were correctly identified as having an association with DILI, whereas the prediction accuracy was as low as 79% when considering the BSEP IC50 values alone (Morgan et al., 2013). However, consideration of the Cmax or Cmax,u values did not improve the predictability of the in vitro assay described here (Supplemental Fig. 5, A and B). A possible reason for this finding is the variable accumulation of each drug inside hepatocytes in vivo (Grime et al., 2008). Alternatively, patients exhibiting DILI might have quite different exposures compared to non-DILI patients. Given this situation, PK parameters determined using non-DILI patients might not improve the predictabilities of assays. Similarly, it was suggested that correction of assay results using PK parameters did not improve the prediction accuracy (Morgan et al., 2013).
We do not have a clear basis for setting the test drug concentration to 50 μM, but this value is empirically chosen based on previous studies (Morgan et al., 2010, 2013; Pedersen et al., 2013; Aleo et al., 2014; Kock et al., 2014). For example, the inhibitory effects of 250 compounds on human BSEP were studied in a membrane vesicle transport assay, and it was found that 86 compounds inhibited human BSEP function significantly (P < 0.05) at 50 μM (Pedersen et al., 2013). To verify the detectability of the test drug at a concentration 50 μM in our SCH-based toxicity assay, the concentration-toxicity relationships in SCRHs were tentatively examined for two selected groups: 1) eight drugs (atorvastatin, clopidogrel, cyclosporine A, flutamide, leflunomide, naftopidil, tacrolimus, and ticlopidine) which showed BA-dependent hepatocyte toxicity at 50 µM, and 2) nine drugs (carbamazepine, clarithromycin, lamivudine, levofloxacin, ranitidine, tranilast, valproate, valsartan, and voriconazole) which did not show BA-dependent hepatocyte toxicity at 50 µM, but their 100 × Cmax,u values are beyond 50 µM. As a result, calculated half-maximal lethal concentration (LC50) values of drugs in the first group were set within the range of 6.0–128 µM, whereas those in the second group were within the range of 183–4380 µM (Supplemental Figs. 6, A and B). It was found that drugs with LC50 < 400 µM apparently showed detectable BA-dependent hepatocyte toxicity in SCRHs when setting the test drug concentration to 50 μM; however, the toxicity of drugs with LC50 < 60 µM seems saturated, whereas the toxicity of drugs with LC50 > 500 µM seems overlooked, implying that drugs with extremely lower or higher LC50 values could not be properly evaluated by fixing the test drug concentration (Supplemental Fig. 6C). From a theoretical point of view, LC50 values would be a more comprehensive index than the cell toxicity data obtained at fixed drug concentration. It is to be determined in a future study if predictability is improved by the use of LC50 values in combination with PK parameters.
Our group also found that efflux transporters other than Bsep might be involved in exporting some BAs from hepatocytes, and that their inhibition aggravates BA-dependent hepatocyte toxicity in SCRHs (Susukida et al., 2015). Flutamide is an example of one such drug that inhibits other efflux transporters. The IC50 of flutamide toward human BSEP and rat Bsep was reported to be as high as 143.2 and 78.7 μM, respectively (Dawson et al., 2012), and 50 μM flutamide did not inhibit human BSEP in a previous study (Pedersen et al., 2013). However, here, flutamide induced BA-dependent hepatocyte toxicity in both SCRHs and SCHHs (Fig. 3), implying the involvement of the inhibition of BA efflux proteins other than BSEP/Bsep. In line with this proposal, it has already been suggested that inhibition assays focusing on BSEP only are insufficient, and the need to consider basolateral efflux transporters such as MRP3 and MRP4 or another unidentified BA transporter for accurate predictions of drug toxicities has been highlighted (Morgan et al., 2013; Kock et al., 2014; Susukida et al., 2015). In fact, the human BSEP inhibition data alone (IC50 values in BSEP-expressed membrane vesicles; see Supplemental Table 1) did not correlate well with abnormal frequencies of serum markers (data not shown).
Other studies categorized DILI risk based on Food and Drug Administration risk classifications, including the black box warning, warning/precaution, adverse reaction, and not mentioned labels. These classifications are not determined solely by the frequency of the marker increase, but also take other factors into account (Avigan, 2014) such as: the estimated rates of life-threatening hepatotoxic adverse events among treated patients; the presence (or absence) of effective tools to reduce DILI risk in treated patients; and regulatory outcomes. Notably, the SCH assay described here simply reflects the probability of BA accumulation inside hepatocytes and subsequent cell death. From this stand point, we think it is reasonable that the assay does not necessarily predict the Food and Drug Administration category classification, but can predict the frequencies of serum test abnormalities (Supplemental Fig. 7). However, some false-negative or false-positive predictions were obtained (Fig. 5). One of the possible reasons is that the in vivo–specific effects—such as the induction of marker enzymes (ALP and γGT)—are responsible for the false-negative cases (e.g., carbamazepine) (Voudris et al., 2005). Moreover, correlation between the frequency of serum bilirubin increase and the frequencies of other serum marker increases in patients is relatively poor (r2 = 0.2581–0.3144) (Supplemental Fig. 1, C–E), and the frequency of serum bilirubin increase was not well correlated with cytotoxicity in both SCRHs and SCHHs, compared with other serum markers (Supplemental Fig. 3, E–H; Supplemental Table 2). One reason to consider concerning why the clinical frequency of serum bilirubin increase did not give good predictability for the risk of cholestatic DILI might be that bilirubin was excreted to the bile by MRP2 but not BSEP (Jedlitschky et al., 1997). Our SCH-based toxicity assay can evaluate the hepatocyte toxicity due to intracellular BA accumulation mainly by BSEP inhibition and, therefore, the serum marker elevated by different mechanisms (i.e., MRP2 inhibition) may not be predicted with our assay system.
In conclusion, BA-dependent cytotoxicity assay using SCHs might be a useful preclinical screening tool to predict the risk of cholestatic DILI. Recently, it was reported that the prediction accuracy of general DILI risk is improved when considering the potential of mitochondrial dysfunction as well as BSEP inhibition (Aleo et al., 2014). These observations are consistent with the understanding that BA-induced apoptosis is one of the major causes of hepatocellular injury (Woolbright and Jaeschke, 2012). From a practical point of view, the assay described here seems beneficial because it does not require knowledge of the clinical dose or target concentration, which are sometimes difficult to estimate during the preclinical drug development stage.
Acknowledgments
The authors express gratitude to LSI Medience Corporation and Shiseido Corporation for gifting cryopreserved human hepatocytes (Hu1437, Hu1524, and Hu4197; Life Technologies).
Authorship Contributions
Participated in research design: Susukida, Sekine, Nozaki, Ito.
Conducted experiments: Susukida, Nozaki, Tokizono.
Contributed new reagents or analytic tools: Susukida, Sekine, Nozaki, Ito.
Performed data analysis: Susukida, Sekine, Nozaki, Tokizono, Ito.
Wrote or contributed to the writing of the manuscript: Susukida, Sekine, Nozaki, Ito.
Footnotes
- Received May 13, 2015.
- Accepted August 31, 2015.
This work was supported by the Japan Society for the Promotion of Science KAKENHI [Grants 24390037 and 23790172], and Leading Graduate School at Chiba University.
The authors state no conflicts of interests.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ALP
- alkaline phosphatase
- ALT
- alanine transaminase
- AST
- aspartate transaminase
- BA
- bile acid
- BSEP
- bile salt export pump
- Cmax,u
- maximum unbound plasma concentration
- DILI
- drug-induced liver injury
- γGT
- gamma glutamyltranspeptidase
- LC50
- half-maximal lethal concentration
- LDH
- lactate dehydrogenase
- MRP
- multidrug resistance-associated protein
- PK
- pharmacokinetic
- PMDA
- Pharmaceuticals and Medical Devices Agency
- ROC
- receiver operating characteristic
- SCH
- sandwich-cultured hepatocyte
- SCHH
- human sandwich-cultured hepatocyte
- SCRH
- rat sandwich-cultured hepatocyte
- TC
- taurocholate
- WME
- Williams’ medium E
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}