Article Text

Abstract
Objective: Case control studies in adults suggest that defective alleles in the gene that codes for the hepatic cytochrome P450 2A6 (CYP2A6) protect against nicotine dependence (ND) and higher levels of cigarette consumption. These two hypotheses were tested in young adolescents.
Design: Self reports of tobacco use and ND symptoms were collected every 3–4 months in a prospective study of 1293 grade 7 students from a convenience sample of 10 schools.
Subjects: 281 smokers with genetic data were analysed; those who were not already tobacco dependent and who had inhaled (n = 228) were followed 29.9 months on average, until they became dependent or were censored.
Main outcome measures: The association between metabolic activity, represented by CYP2A6 genotype, and conversion to dependence was analysed using Cox’s proportional hazards model.
Results: During follow up 67 subjects (29.4%) became dependent. Relative to CYP2A6*1/*1, having 1–2 copies of the inactive CYP2A6*2 or *4 variant was a strong risk factor for developing dependence (hazard ratio 2.8, 95% confidence 1.3 to 6.3). Subjects with 1–2 partially inactive CYP2A6*9 or *12 variants were not at increased risk. Mean past-week cigarette consumption at the end of follow up (controlling for age, sex, and number of months since first inhalation) among dependent subjects was 29.1 among normal inactivators, compared to 17.2, and 12.7 among slower (1–2 copies of *9 or *12), and slowest (1–2 copies of *2 or *4) inactivators, respectively (p = 0.09).
Conclusion: Adolescents with 1–2 copies of CYP2A6*2 or *4 are at substantially increased risk of becoming dependent but smoke less once dependent. Genetic risk for ND may need to be considered in the conceptualisation of tobacco control programmes for adolescents.
- CYP2A6
- adolescent
- nicotine dependence
- smoking onset
- tobacco use
Statistics from Altmetric.com
The search for determinants of tobacco use initiation and maintenance, the development of nicotine dependence (ND), and levels of cigarette consumption has broadened over the last five years to include a variety of candidate genes, in addition to sociodemographic, psychosocial, and environmental risk factors.1 This search is motivated by the belief that information on genetic predisposition and eventually gene–environment interactions will help tailor prevention and cessation interventions to individual need.
Genes involved in the metabolism of nicotine are biologically plausible candidates for studies of smoking behaviour.2,3 In particular, there is considerable inter-individual variation in levels of the genetically polymorphic hepatic enzyme CYP2A6,2,4 which inactivates 80–95% of nicotine to cotinine.5,6 Seventeen CYP2A6 genetic variants have been identified to date including CYP2A6*1-*16 and the gene duplication, *1×2.4,7 Only a few of these variants, however, have been demonstrated to alter enzymatic function in vivo. The CYP2A6*2 allele which contains a detrimental point mutation, and CYP2A6*4 which contains a gene deletion, are both fully inactive.2,4 Others including CYP2A6*9 and CYP2A6*12 result in decreased enzyme activity due to decreased transcription rates and altered enzyme structure, respectively.8,9
One hypothesis of interest in regard to nicotine metabolism is that slow inactivators of nicotine (that is, individuals with genetic variants of CYP2A6) may be less likely to maintain smoking once they have initiated, and thus have a lower risk of developing ND. This is based on the hypothesis that there is little tolerance to the psychoactive effects of nicotine during early onset such that, for the same amount of nicotine inhaled, slow inactivators may experience higher or longer lasting levels of nicotine. This could increase symptoms of nicotine toxicity such as dizziness and nausea, thereby reducing the likelihood of maintaining smoking.10 Several case control studies in adults provide support for an association between genetic impairment of CYP2A6 and decreased risk for being tobacco dependent,10–14 although others do not.15–17
A second hypothesis of interest is that once ND is established, slow inactivators may require fewer cigarettes to maintain nicotine concentrations at optimal levels. Several studies suggest that dependent smokers adjust their smoking to maintain nicotine levels.18,19 More specifically, smoking is increased if the nicotine content of cigarettes is decreased or if nicotine excretion is increased by urine acidification; smoking is decreased with concurrent intravenous or patch nicotine.2 Again several,13,20,21 but not all14,16,17 studies support the notion that slow nicotine inactivators smoke fewer cigarettes.
In this current analysis of data from a prospective cohort study of young adolescents, we tested the hypothesis that partially or fully inactive CYP2A6 variants protect against early development of tobacco dependence in novice smokers. We also examined early smoking symptoms to determine if slow inactivators experience dizziness and nausea more frequently than normal inactivators, when they first begin to smoke. Finally, we examined if partially or fully inactive CYP2A6 variants protect against higher levels of cigarette consumption.
METHODS
The McGill University Study on the natural history of nicotine dependence in teens (NDIT study) is an ongoing prospective study of 1293 students recruited in fall 1999 from all grade 7 classes in a convenience sample of 10 secondary schools in Montreal. The primary objectives are to study the onset of ND symptoms in relation to first tobacco exposure, and to investigate the relative etiologic importance of environmental and genetic risk factors for ND. Secondary schools were selected to include a mix of French and English schools, urban, suburban, and rural schools, and schools located in high and low socioeconomic neighbourhoods. All subjects and a parent/guardian provided written, informed consent. Over half (55.4%) of eligible students participated in the baseline data collection; the low response is related in part to the need for a blood draw for genetic analysis, and also to a labour dispute in Quebec schools during baseline data collection which affected the collection of consent forms. The McGill University Faculty of Medicine Institutional Review Board approved the study protocol.
Data collection comprised self report questionnaires administered at school every 3–4 months during the school year. A blood draw for DNA analysis was completed in March 2002; 561 of 1054 eligible subjects (53.2%) provided written parental consent for the blood draw. Blood specimens were available for 523 subjects, of whom 281 (53.7%) were white (we restricted the DNA analysis to white subjects) and had initiated cigarette smoking either before the first wave of data collection (hereafter called “baseline smokers”) or during follow up (hereafter called “initiators”).
Lifetime smoking history was determined at each follow up in two items adapted from previous research.22 Six items measured smoking in a three month recall beginning with the month preceding questionnaire administration23; one item for each month measured the number of days on which the subject had smoked during the month, and one item for each month measured the number of cigarettes smoked per day on average during that month. The number of cigarettes smoked during the week preceding questionnaire administration was measured in a seven day recall.24 Age at first inhalation was ascertained among subjects who reported that they had inhaled.
Early smoking symptoms of nicotine toxicity were assessed by asking subjects whether they had experienced each of dizziness and nausea a lot, a bit, or not at all the first few times they took cigarette smoke into their lungs. Data for these variables were drawn from the first questionnaire completed among baseline smokers, and from the first questionnaire in which the subjects reported inhaling among initiators.
Our measure of tobacco dependence included 2–4 items for each of the six criteria of the International classification of diseases, 10th revision (ICD-10) definition of tobacco dependence, for a total of 18 items.25 Each item had multiple response choices; an item was considered positive only if the most extreme positive response choice was endorsed. A criterion was considered positive if any of its items was positive (the withdrawal syndrome required that two or more of four items be endorsed). A subject was categorised as tobacco dependent (yes, no) if he/she met three or more of the six ICD-10 criteria. In previous work,26 internal reliability and two week test retest reliability of the ICD-10 measure were excellent (Cronbach’s α = 0.91, 100% test retest agreement), and in addition the measure showed strong evidence of convergent construct validity.
Genotyping
Genotyping was performed for five variant alleles using established methods based on gene and allele-specific two step polymerase chain reaction (PCR) assays.4,9,11 The variants investigated were selected based on their impact on nicotine metabolism and on reported frequencies in whites.4 The analyses identified two partially inactive variants (CYP2A6*9, *12), two fully inactive variants (CYP2A6*2, *4), and the increased activity gene duplication, CYP2A6*1x2.
Data analysis
The sample included 281 white baseline smokers and initiators with venepuncture. Subjects were categorised into three groupings according to genotype including normal inactivators (two copies of *1, the wild type active allele), slower inactivators (1–2 copies of *9 or *12, or one copy each of *9 and *12) and slowest inactivators (1–2 copies of *2 or *4). Data for this analysis were drawn from the first four years of follow up.
To estimate the incidence of conversion to tobacco dependence, subjects were followed from time of first inhalation until they became dependent or until they were censored (lost to follow up or study period ended). The association between the rate of conversion to dependence and the presence of defective relative to normal variants was examined in Kaplan-Meier actuarial analysis. Hazard ratios for the development of dependence for each genotype category relative to the CYP2A6*1/*1 genotype were derived from Cox’s proportional hazards model. Because they were more comparable with respect to bias related to recall of age at first inhalation, ever smokers and initiators were analysed in separate strata.
To examine if bias related to recall of age at first inhalation affected parameter estimates, we restricted a second analysis to subjects who reported their first inhalation after the baseline. Subjects were followed from the time of first inhalation until they converted to dependence or until they were censored.
Role of the funding source
The study sponsor had no role in the study design, collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
RESULTS
Overall 12.8% of subjects had one or two copies of partially inactive CYP2A6 variants *9 or *12; 6.4% had one or two copies of inactive variants *2 or *4 (table 1). There were no important differences in the distribution of these variants by sex, age, or language. Hardy-Weinberg computations indicated that the observed genotype distribution was not different from the expected distribution, indicating no evidence of non-random selection.
Distribution of genotype according to metabolic activity among all subjects, those who had inhaled, and those who initiated inhalation during follow up
There were fewer smokers among subjects with venepuncture than among those without venepuncture (28.2% v 35.1%). Compared to never smokers with venepuncture, there were higher proportions of females (59.8% v 42.6%), of Francophones (33.0% v 24.0%), of subjects whose mothers smoked (32.4% v 12.0%), and of subjects whose fathers smoked (30.8% v 18.7%) among ever smokers with venepuncture. table 2 shows selected characteristics of subjects included in the current analysis. The prevalence of smoking among parents of subjects with at least one copy of inactive CYP2A6 variants was notably lower than among parents of other subjects. Contrary to the study hypothesis, slow inactivators did not report more early smoking symptoms of dizziness or nausea than normal inactivators (table 2).
Comparison of selected characteristics of normal, slower, and slowest inactivators (n = 281)
Fifty three subjects were not at risk of developing tobacco dependence either because they were already dependent at baseline (n = 7) or because they had never inhaled (n = 46); these subjects were excluded from further analysis. Ten of the 228 subjects retained for further analysis were lost to follow up; one subject dropped out and nine moved.
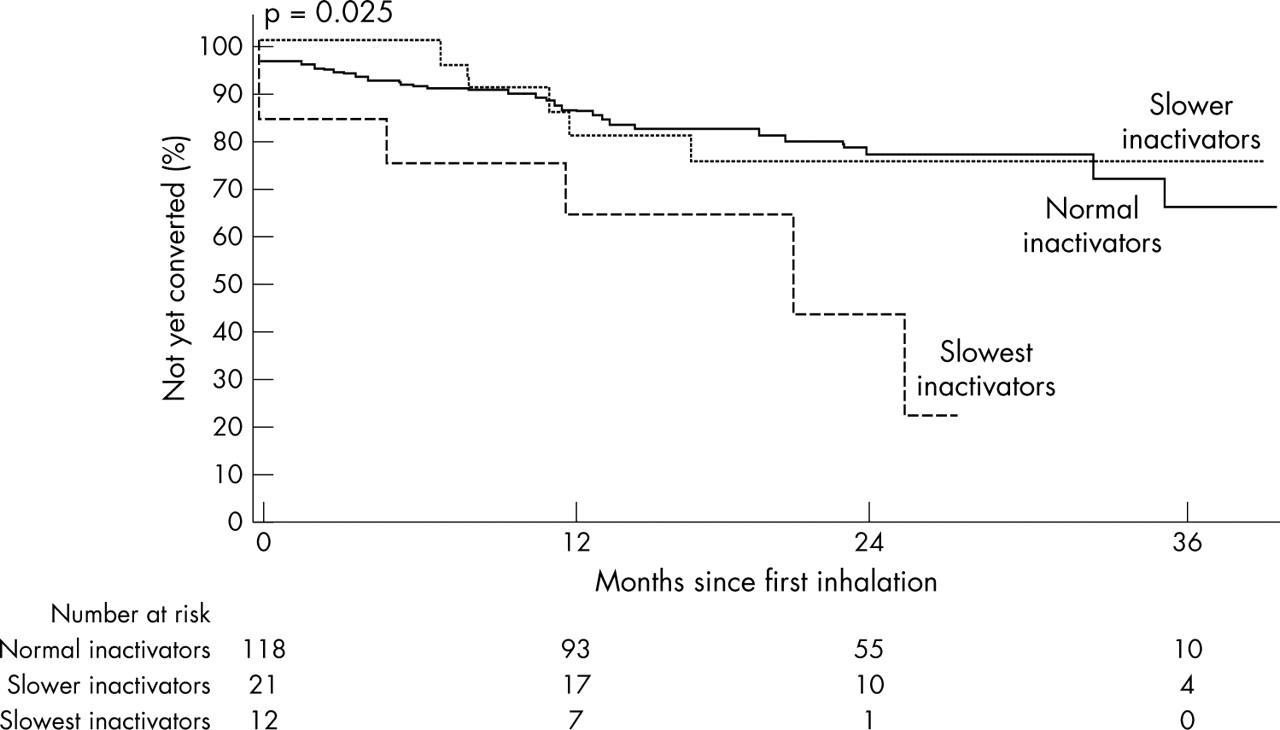
Over an average 29.9 (SD 20.7) month follow up from time of first inhalation, 67 of 228 subjects (29.4%) became tobacco dependent. The incidence of conversion was substantially higher among the slowest inactivators, compared to other subjects (table 3). Figure 1 compares Kaplan-Meier curves according to genotype. The Cox’s proportional hazard ratios confirmed the Kaplan-Meier analysis and suggested that the risk of conversion was almost three times higher among the slowest inactivators relative to normal inactivators (hazard ratio (HR) 2.8, 95% confidence interval (CI) 1.3 to 6.3) (table 3).
Incidence and hazard ratio of conversion to tobacco dependence according to metabolic activity

Actuarial estimates of conversion to ICD-10 tobacco dependence according to metabolic activity among subjects who had inhaled (n = 228). The p value was calculated with the use of the log rank test.
In an analysis restricted to the 151 subjects who initiated inhalation after the baseline data collection (table 3, fig 2), the risk of conversion for the slowest inactivators remained significant (HR 3.2, 95% CI 1.3 to 7.7).

{kind=link}
{kind=link}
Actuarial estimates of conversion to ICD-10 tobacco dependence according to metabolic activity among subjects who initiated inhalation during follow up (n = 151). The p value was calculated with the use of the log rank test.
To test if the partially inactive or inactive CYP2A6 variants protect against higher levels of cigarette consumption, we compared past week cigarette consumption at the end of follow up among dependent and non-dependent subjects by genotype, controlling for age, sex, and months since first inhalation. There was a non-significant trend toward lower cigarette consumption among slowest and slower inactivators who were dependent compared with dependent normal inactivators (12.7, 17.2, and 29.1 cigarettes in the past week, respectively (table 4); median = 11.5, 16, and 21 cigarettes in the past week).
Past week cigarette consumption at the end of follow up among subjects who initiated inhalation during follow up, according to ICD-10 dependence and metabolic activity
DISCUSSION
Recent studies suggest that early ND symptoms play a central role in novice smokers maintaining the smoking habit and becoming addicted to tobacco,27,28 and it is well established that ND is associated with smoking cessation failures. However, although there is substantial genetic risk for several aspects of smoking, including the development of ND and the amount smoked,1 no studies to date have investigated genetic risk prospectively in novice smokers.
Defective CYP2A6 alleles are promising candidate determinants from a biological perspective, although case–control studies of adults yield conflicting results. Inherent difficulties in these studies including bias related to population stratification, cases and controls drawn from different populations, crude phenotype assessments, undetermined co-morbidity, variable genotyping methodologies, examination of different variant alleles, and poor recall of smoking history, cigarette consumption, and symptoms of nicotine dependence, could explain the lack of replication.3 In this current study, we minimised recall bias by studying conversion to dependence in a prospective study of healthy young novice smokers, and we reduced misclassification bias by using a validated measure of tobacco dependence with strong psychometric properties. Although this is the first study of CYP2A6 in adolescents there is little reason to suspect that nicotine metabolism, or the effects of variant and non-variant alleles in this age group, differ from that observed in adults. Gourlay and Benowitz29 reported no differences in steady state nicotine plasma concentrations or estimated plasma clearance values in three age groups (18–39, 40–59, and 60–69 years) following application of nicotine patches. The half life of cotinine, which is metabolised exclusively by CYP2A6, is similar in neonates, older children, and adults.30,31 Finally other CYP2A6 substrates such as coumarin and caffeine are metabolised similarly in children of different ages and adults.32–34
Despite the evidence in adults, none of the defective CYP2A6 variants investigated in this study protected subjects from developing dependence. In contrast having 1–2 copies of an inactive variant increased the risk substantially. It thus seems possible that slow nicotine inactivation results in prolonged and/or higher brain exposure to nicotine, and that this more intense exposure might enhance the neurophysiological processes that lead to dependence. It is possible that even very brief exposures to nicotine during adolescence can result in long term neurological adaptations. Abreu-Villaca et al35 demonstrated an increase in the number of nicotinic acetylcholine receptors in the hippocampus of adolescent rats in response to four injections of nicotine, which is equivalent to smoking two cigarettes on two consecutive days. We postulate that genetic alterations that slow nicotine metabolism increase the likelihood that the first few cigarettes smoked by an adolescent will initiate the neurological alterations that lead to dependence.
In addition to difficulties in the case–control design, another explanation for our findings relative to adult case–control studies is that genetically slow nicotine metabolism has different effects at different stages of smoking. It may be detrimental by increasing the risk for becoming dependent in novice smokers, but beneficial by increasing the ability to quit smoking in established smokers. In fact the proportion of slow metabolisers among adult current smokers decreases with duration of smoking,11 suggesting that slow metabolisers might quit more readily than normal metabolisers. Gu et al13 reported that white adults with CYP2A6*2 alleles smoked for a shorter duration and were 1.75 (95% CI 1.17 to 2.61) times more likely to quit relative to adults with no CYP2A6*2 allele. Similar findings have also been observed among African Americans.36 Finally, slower metabolisers in this current study smoked fewer cigarettes per week, and lower cigarette consumption has been associated in studies of adults with better cessation outcomes.37,38 It is possible that under-representation of adult smokers with CYP2A6 genetically mediated slow nicotine inactivation who have quit smoking (selection bias), or possibly misclassification of smokers who have quit, results in an apparent protective effect of slow metabolism in adult case–control studies. Our observation of a lower prevalence of smoking among parents of slow inactivators is consistent with this reasoning.
According to our hypotheses, symptoms associated with early smoking act in the causal pathway between genotype and tobacco dependence. Because of a presumed aversive reaction to these symptoms, subjects with defective alleles were hypothesised to avoid cigarettes, thus minimising the risk of becoming dependent. Our data do not support the hypothesis that higher circulating concentrations of nicotine represented by genotype are associated with increased early smoking symptoms of dizziness or nausea. Therefore it is unlikely that, even if defective alleles were protective against developing ND, this association is mediated through symptoms associated with early smoking.
Consistent with our observations in this study, previously reported prevalences of CYP2A6*9 and *12 alleles are 5.2% and 2.2%, respectively, in whites.4,9,11 Similarly approximately 6% of study subjects in this sample had a genotype which included at least one copy of the CYP2A6*2 or *4 variant. The population attributable risk (PAR% = p (RR–1)/(p (RR–1)+1) ×100) related to the CYP2A6*2 or *4 variants among white adolescent ever smokers is therefore in the range of 14.6%, indicating that this genetic predisposition is a public health issue that warrants attention.
Previous reports suggest that defective CYP2A6 variants protect against heavy tobacco use.11,13,20,21 Our data show that cigarette consumption at the end of follow up was notably lower among dependent subjects with reduced activity alleles. This is consistent with the notion that slow metabolism results in sustained high exposure to nicotine and therefore subjects with defective alleles require fewer cigarettes to maintain nicotine concentrations at optimal levels.
Overall these data provide compelling evidence for genetic influences in the development of ND and possibly levels of cigarette consumption. However, because most novice smokers are intermittent smokers with low cigarette consumption, we need to better understand how defective CYP2A6 genotypes affect peak nicotine concentrations after a single cigarette, and how long nicotine remains after smoking a single cigarette. As suggested above, it is possible that following the initial phase of intake of nicotine from a single cigarette, the plasma and brain concentrations of nicotine decrease more slowly in slow metabolisers resulting in higher and longer retention of nicotine in blood and brain concentrations. If a second cigarette is smoked within hours the peak blood concentrations could be higher due to the higher blood concentrations that exist before the second cigarette is smoked. There is also a need for continued assessment of adolescents through the later stages of smoking. More specifically, the stage of smoking at which slow nicotine inactivation might begin to confer an advantage with respect to quitting should be determined.
What this paper adds
Several, although not all, case–control studies in adults provide support for an association between genetic impairment of CYP2A6 and decreased risk for being tobacco dependent. However, inherent difficulties in the case–control design include bias related to population stratification, cases and controls drawn from different populations, crude phenotype assessments, undetermined co-morbidity, variable genotyping methodologies, examination of different variant alleles, and poor recall of smoking history, cigarette consumption, and symptoms of nicotine dependence. In this prospective study of novice smokers, we showed that adolescents with 1–2 copies of CYP2A6*2 or *4 are at substantially increased risk of becoming dependent, raising concerns that genetic risk for nicotine dependence may need to be considered in the conceptualisation of tobacco control programmes for adolescents.
Study limitations
The subjects retained for analysis represent a distinct subgroup of the larger sample. It included (1) only white subjects, (2) only subjects who consented and participated in the blood draw; and (3) among subjects with blood draw, only those who had initiated smoking. We did not genotype all subjects with blood draw because of budget restrictions. While subjects retained for analysis represent fewer that 25% of the sample, it is unlikely that this biased the main findings on the association between genotype and onset of dependence—refusal to participate in the blood draw (or in the overall study) is not likely to relate to genotype because all potential participants were unaware of their CYP2A6 status.
The sample size of subjects with the inactive CYP2A6*2 or *4 variants who became tobacco dependent was small, but this did not impede detection of a significant association between these variants and conversion to tobacco dependence. Data on ICD-10 symptoms of tobacco dependence were drawn from self reports by adolescents. However, our ICD-10 indicator has been shown to be internally reliable, to have excellent test retest reliability, and to show convergent construct validity.26 Although the proportion of subjects with blood draw was modest, there were few meaningful significant differences between subjects retained and those not retained for this analysis. In addition, because loss to follow up was minimal, there is little reason to suspect that selection bias affected the results, or that external generalisability is an issue of concern.
Conclusion
Adolescents with slowed nicotine metabolism due to genetic defects in CYP2A6 are at substantially increased risk of becoming tobacco dependent at relatively low levels of cigarette consumption. The role that genetically mediated slow nicotine inactivation may have at different stages of smoking (that is, development of dependence, success in cessation) needs to be clarified. Consistent with adult studies, adolescents with slow nicotine inactivation smoke less than normal inactivators. Overall these data suggest that genetic risk for ND should be taken into account in the conceptualisation of prevention and cessation programmes for adolescents.
Acknowledgments
This research was funded by the National Cancer Institute of Canada with funds from the Canadian Cancer Society. J O’Loughlin is an Investigator of the Canadian Institutes of Health Research. We acknowledge the support of a Canadian Institutes of Health Research Tobacco Research Training award to W Kim and a Canadian Research Chair in Pharmacogenetics to RF Tyndale. The genotyping was funded in part by CIHR grant MOP 53248 to RF Tyndale and by the Centre for Addiction and Mental Health. The authors thank G Chong for the DNA extraction, PBS Clarke for comments on the manuscript, and B Xu for invaluable technical support.
REFERENCES
Footnotes
-
↵* Also Direction de santé publique de Montréal-Centre, Montréal, Québec, Canada