Article Text

Abstract
Objective: To examine the therapeutic activity of hydrophilic glucocorticoid encapsulated in PLGA nanoparticles, which have shown slow release and are targeted to inflamed joints after intravenous administration, in experimental arthritis models.
Methods: Betamethasone sodium phosphate (BSP) encapsulated in PLGA nanoparticles with a size of 100–200 nm (PLGA-nanosteroid) was prepared using a modified oil in water emulsion solvent diffusion method with Zn ions and coated with lecithin. Rats with adjuvant arthritis (AA rats) and mice with anti-type II collagen antibody induced arthritis (AbIA mice) were treated intravenously with PLGA-nanosteroid after the initial sign of arthritis.
Results: In AA rats, a 30% decrease in paw inflammation was obtained in 1 day and maintained for 1 week with a single injection of 100 μg of PLGA-nanosteroid. Soft x ray examination 7 days after this treatment showed decreased soft tissue swelling. Moreover, the PLGA-nanosteroid was also highly effective in AbIA mice. A single injection of 30 μg of the PLGA-nanosteroid resulted in almost complete remission of the inflammatory response after 1 week. In contrast, the same dose of free BSP after three administrations only moderately reduced the severity of inflammation. In addition, a histological examination 7 days after the treatment showed a significant decrease of the inflammatory cells in the joints.
Conclusion: The observed strong therapeutic benefit obtained with PLGA-nanosteroid may be due to the targeting of the inflamed joint and its prolonged release in situ. Targeted drug delivery using a sustained release PLGA-nanosteroid is a successful intervention in experimental arthritis.
- AA, adjuvant induced arthritis
- AbIA, antibody induced arthritis
- BSP, betamethasone sodium phosphate
- IP, intraperitoneally
- IV, intravenously
- LPS, lipopolysaccharide
- PLA, poly(d, l-lactic acid)
- PLGA, poly(d, l-lactic/glycolic acid)
- RA, rheumatoid arthritis
- poly(d, l-lactic/glycolic acid)
- nanoparticles
- betamethasone sodium phosphate
- adjuvant induced arthritis
- rats
- antibody induced arthritis
- mice
Statistics from Altmetric.com
- AA, adjuvant induced arthritis
- AbIA, antibody induced arthritis
- BSP, betamethasone sodium phosphate
- IP, intraperitoneally
- IV, intravenously
- LPS, lipopolysaccharide
- PLA, poly(d, l-lactic acid)
- PLGA, poly(d, l-lactic/glycolic acid)
- RA, rheumatoid arthritis
- poly(d, l-lactic/glycolic acid)
- nanoparticles
- betamethasone sodium phosphate
- adjuvant induced arthritis
- rats
- antibody induced arthritis
- mice
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterised by joint synovial inflammation and progressive cartilage and bone destruction, but its pathogenesis is not yet clearly understood.1
Glucocorticoids can be highly effective in treating joint inflammation, but their systemic application is limited because of a high incidence of serious adverse effects, especially related to long term treatment.2,3 As intravenously administered glucocorticoids are distributed throughout the whole body and rapidly cleared, high and frequent dosing is necessary to achieve an effective concentration at inflamed target sites. Moreover, the profound physiological activity of glucocorticoids in many different tissues increases the risk of adverse effects in patients. It is necessary, therefore, to develop a drug delivery system for glucocorticoids incorporated in particular carriers, with enhanced localisation in the target site and sustained drug release.4,5
Because the long-circulating liposome system for drug delivery had already been introduced into clinics,6,7 a report has recently been published using polyethylene glycol-liposome containing prednisolone phosphate for the treatment of arthritis.8 However, the allergic reaction against liposome preparations even in the pegylated form becomes a problem to overcome.9 Meanwhile many researchers have developed poly(d, l-lactic/glycolic acid) (PLGA) or poly(d, l-lactic acid) (PLA) microparticles and nanoparticles that encapsulate water insoluble, low molecular weight pharmaceutical agents, which are employed for clinical use.10–,13 We also have developed nanoparticles, in which a water soluble, non-peptide, low molecular weight pharmaceutical agent is made hydrophobic by a metal ion and encapsulated in nanoparticles formed of PLGA/PLA, with a surfactant applied to the surface of nanoparticles with a diameter of 100–200 nm. They can target a specific lesion after intravenous administration and are less likely to burst at an early stage of administration so that they can gradually release the agent at the site of the lesion over a prolonged period of time (J Controlled Release, in press).
PLGA-nanosteroid for intra-articular preparation has been reported,14,15 but the encapsulating rate of steroids is low and their size is not suitable for intravenous administration. We therefore prepared a PLGA-nanosteroid for intravenous preparation by efficiently encapsulating betamethasone sodium phosphate (BSP) by a modified oil in water solvent diffusion method as described before.
We studied the therapeutic activity of the intravenous PLGA-nanosteroid in arthritis models of rats with adjuvant arthritis (AA)16,17 and mice with type II collagen antibody induced arthritis (AbIA)18–,20 to evaluate whether this preparation might improve the delivery and therefore the efficacy of glucocorticoids.
MATERIALS AND METHODS
Preparation of the PLGA-nanosteroid
We prepared the PLGA-nanosteroid as described before (J Controlled Release, in press). In brief, PLGA with a lactic acid/glycolic acid ratio of 50/50 (mol wt 13 000) was purchased form Wako Pure Chemicals Industries, Ltd (Osaka, Japan). BSP and lecithin were purchased from Sigma Chemical Co (St Louis, MO, USA). BSP loaded nanoparticles were prepared using the modified emulsion solvent diffusion method in oil. One millilitre of 0.5 M zinc acetate (Wako) was added to 4 mg of BSP in 500 μl of deionised distilled water (DDW) and the pellets were collected. After centrifugation at 20 000 g for 10 minutes, the pellets were dissolved in 0.6 ml of acetone with 20 mg of PLGA (mol wt 8000), and 10 μl of 0.5 M zinc acetate was added. Then, 5 ml of dispersing DDW containing 0.5% lecithin was added and purified by ultrafiltration (YM-50, Millipore Co, Billerica, MA.), after gel filtration with a PD-10 column (Amersham Bioscience, Tokyo, Japan).
Animals
Lewis rats (7 week old males, weighing 200–250 g) and BALB/c mice (6 week old females, weighing 20–25 g) obtained from SLC (Shizuoka, Japan) were used for this study. The animals were housed in a specific pathogen free environment and allowed free access to food and water. The Institution Committee of Animal Experiments in St Marianna Medical University approved all animal studies.
Induction of adjuvant arthritis
Arthritis was induced in Lewis rats by injecting 50 μl of incomplete Freund’s adjuvant solution (DIFCO, Detroit, Mich) containing 6 mg/ml of Mycobacterium butyricum, under ether anaesthesia, into the subplantar region of the left hind paw.21,22 Fourteen days after administration of the adjuvant, when the joint inflammation for all rats reached a maximum in the experiment, the animals were divided into groups (seven in each) so that there were no significant differences between the groups in the volume of the left hind leg of the animals. In addition, a single dose of PLGA-nanosteroid, blank PLGA nanoparticles, or saline was administered intravenously to each group. As controls, a single dose of BSP (100 μg or 300 μg) was subcutaneously administered to the respective groups of rats (seven in each). The development of arthritis in the left hind paw was monitored plethysmographically by recording changes in paw volume by water displacement with model TK-101 (Muromachi Co Ltd, Tokyo). The inflammation rate was calculated with the following equation:
Inflammation rate (%) = (measured leg volume−leg volume without adjuvant)/(leg volume on day 14−leg volume without adjuvant)×100.
The average (SD) of the leg volume without the adjuvant is 1.5 (0.2) ml. Body weights were also monitored daily during the experiment.
The swelling in the paw was assessed by a soft tissue x ray examination on day 21.23 Normal and arthritic rats with the treatment were anaesthetised with sodium pentobarbital (45 mg/kg, intraperitoneally (IP)), placed on a radiographic box, and radiographs of the hind paws were obtained with a Philips ×12 machine (40 kW for 0.01 seconds).
Induction of antibody induced arthritis
Arthritis was induced using the methods of Terato et al.18 Balb/c mice were injected intravenously with 2 mg/kg of an arthritogenic monoclonal antibody cocktail (Chondrex, LLC, Seattle, WA) on day 0, and lipopolysaccharide (LPS; 2.5 mg/kg) was intraperitoneally injected on day 3. This protocol induces severe arthritis within 48 hours after the LPS injection and persists for more than 1 week. Intravenous treatment with a PLGA-nanosteroid (100 μg ×1), BSP (100 μg in phosphate buffered saline, once or three times daily), blank PLGA nanoparticles, or saline (seven in each group) was started 2 days after LPS administration (on day 5). Mice were weighed daily and examined for visual signs of inflammation. The clinical severity was graded as follows: 0, normal; 1, erythema; 2, slight swelling; and 3, severe swelling or deformity. Each limb was graded, allowing a maximum clinical score of 12 for each animal.24
On day 12, animals were anaesthetised with sodium pentobarbital (45 mg/kg, IP) and killed. The distal one third of the limbs was immersion fixed in formalin, decalcified in HCl, processed by routine methods, and embedded in paraffin. The specimens were sectioned at 4–6 μm, stained with haematoxylin and eosin, and examined by light microscopy.19,20 The extent of cellular infiltration and the erosion of join cartilage was separately determined and graded from 0 to 3 (3 is the severest) in each paw.
Statistics
The Mann-Whitney U test was conducted for statistical analysis. Values of p<0.05 were considered significant.
RESULTS
AA rats
Three days after administration of the adjuvant, the first signs of joint inflammation became apparent (paw volume; 3.0 (0.2) ml), in conjunction with a loss of body weight due to the onset of adjuvant induced arthritis. Fourteen days later, the disease reached its maximum severity (paw volume 4.8 (0.3) ml), after which the inflammation gradually resolved. Although vehicle-PLGA nanospheres and saline did not show any anti-inflammatory effect, the PLGA-nanosteroid exhibited, as early as 1 day after administration, a high anti-inflammatory effect (62 (3)%) and continued to show the effect over a succeeding 7 days; meanwhile the anti-inflammatory effect of free BSP was gradually lost over time (fig 1⇓). The anti-inflammatory effect of the PLGA-nanosteroid was better than that of the vehicle control (p<0.01, days 15–21) and the 300 μg dose of free BSP (p<0.05, days 16–21). There was no difference in body weight change among the four groups.

Paw inflammation rate of AA rats. Arthritis was induced in Lewis rats as described in the “Materials and methods” section. Inflammation rate (%) = (measured leg volume−leg volume without adjuvant)/(leg volume on day 14−leg volume without adjuvant)×100. The average inflammation rate of seven rats from days 14 to 21 in each group is shown. SEM is within 10%. *p<0.05 (PLGA-nanosteroid v 300 μg BSP); †p<0.01 (PLGA-nanosteroid v vehicle-PLGA nanoparticles).
In the soft x ray examination, a marked reduction of swelling in the left paw was seen in the PLGA-nanosteroid treated rats (fig 2⇓). Cartilage erosion could not be prevented completely in this experiment, because the treatment was started after the maximum severity of arthritis.

Results of soft tissue x ray examination of the hind paw of AA rats. The swelling in the paw was assessed by soft tissue x ray examination on day 21 as described in “Materials and methods”. (A) Normal rat control; (B) PLGA-nanosteroid (100 μg, intravenously (IV)) treated AA rat; (C) BSP (100 μg) treated AA rat; (D) vehicle-PLGA nanoparticle treated AA rat.
AbIA mice
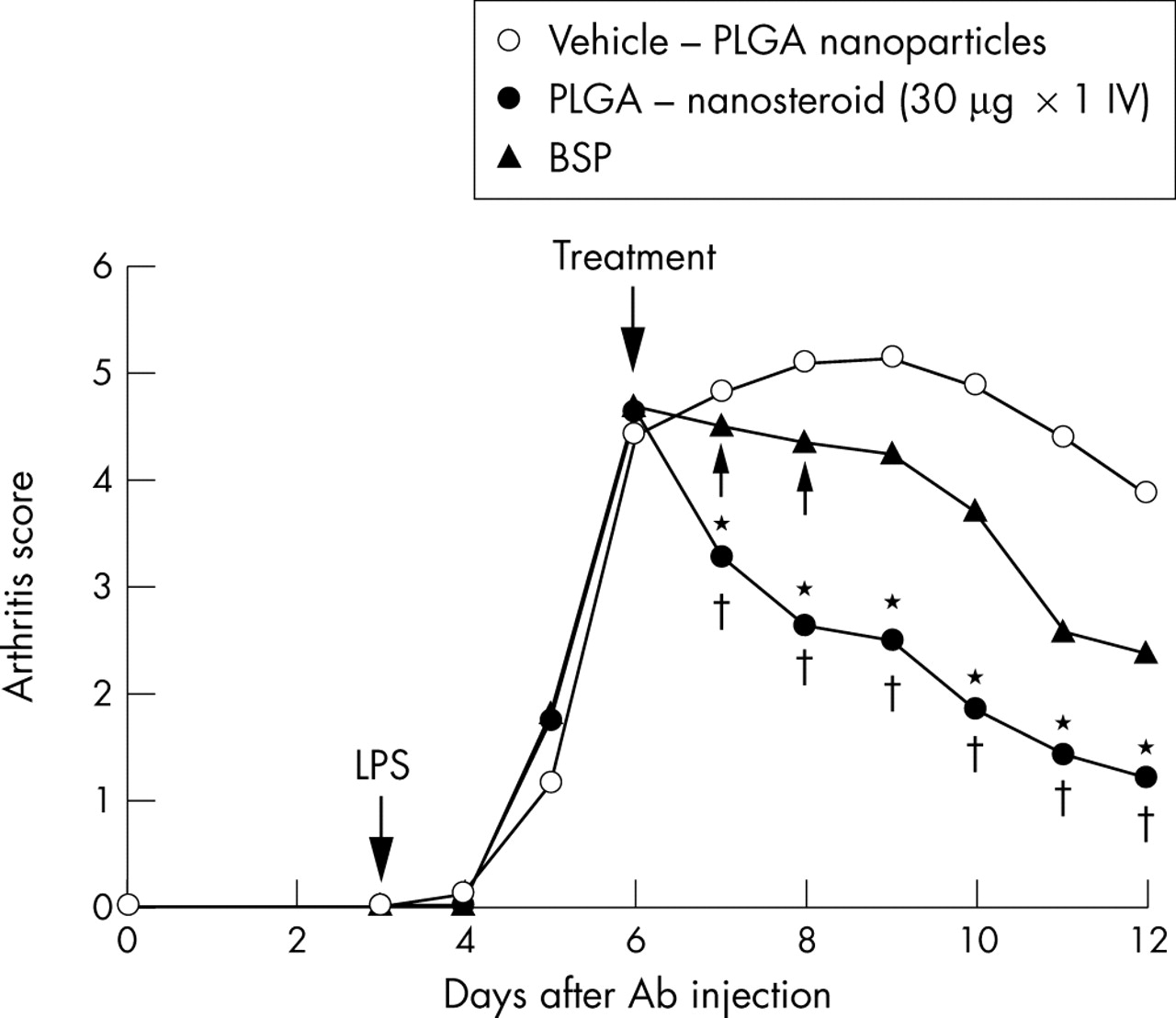
AbIA developed rapidly in immunised mice, and clinical signs (periarticular erythema and oedema) of the disease first appeared in the front paws 2 days after the LPS challenge (on day 5) with a 100% incidence. Erythema and swelling in the hind and front paws increased in frequency and severity in a time dependent manner, reaching maximum arthritis indices of 5.5 (0.2) on day 7 in the vehicle treated mice. The clinical score of the vehicle treated mice was as severe as that of the saline treated mice (data not shown). In contrast, PLGA-nanosteroid treated mice on day 7 had macroscopic evidence of disease with a reduced score of 3.8 (0.1) on day 7 (p<0.01). In addition, PLGA-nanosteroid treated mice demonstrated a significant reduction in joint inflammation over the succeeding 5 days (*p<0.05 v three doses of BSP; †p<0.01 v vehicle control) (fig 3⇓).
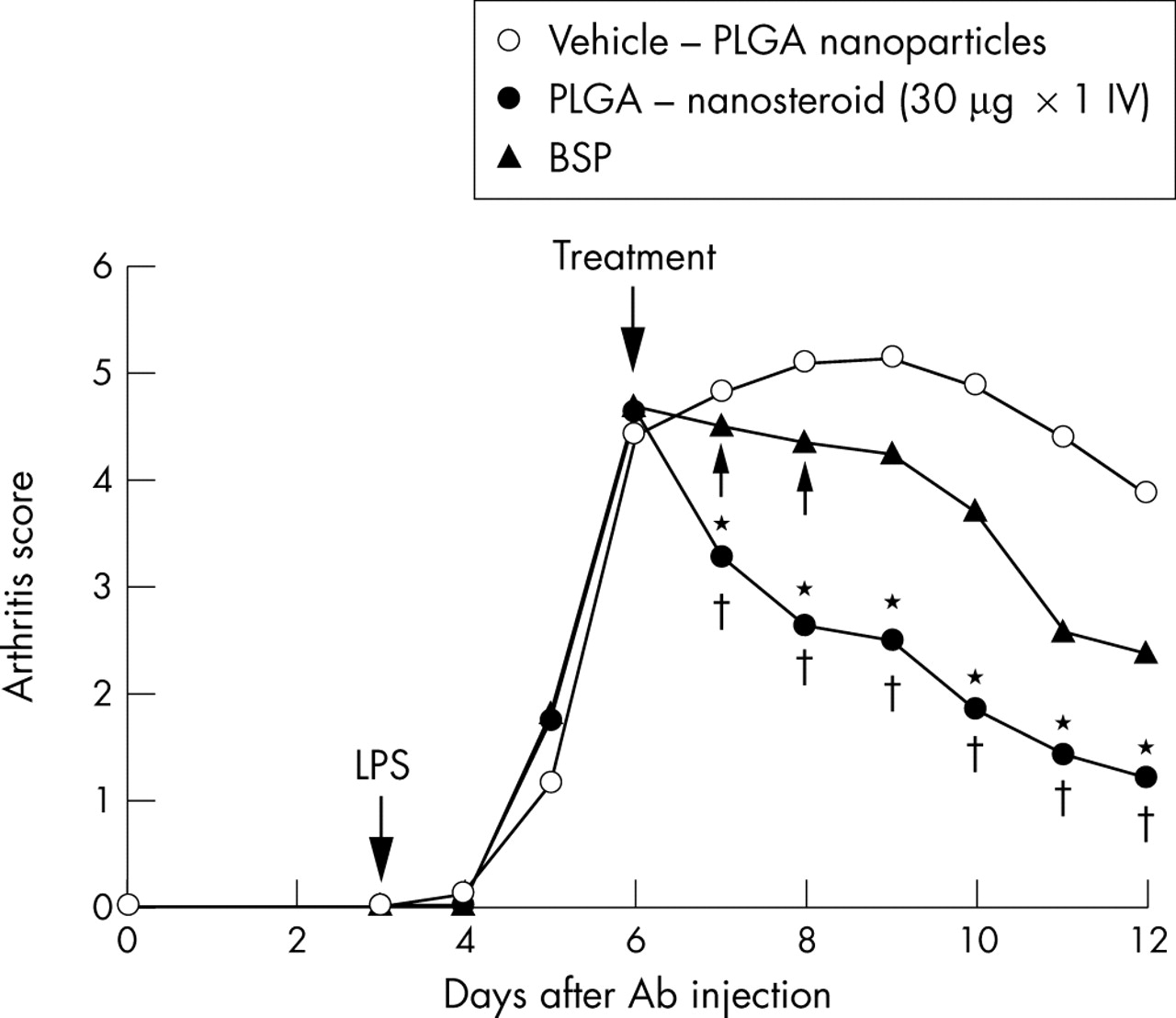
Paw inflammation score of AbIA mice. Arthritis was induced in Balb/c mice as described in the “Materials and methods” section. The average clinical score is shown. SEM is within 10%. *p<0.05 (PLGA-nanosteroid v BSP) ; †p<0.01 (PLGA-nanosteroid v vehicle-PLGA nanoparticles).
Histological examination on day 12 showed that treatment with PLGA-nanosteroid completely inhibited the influx of inflammatory cells, while a severe inflammatory reaction was seen in the control animals (fig 4⇓). There was no macroscopic evidence of either paw erythema or oedema in the sham treated mice. The rate of incidence and the absolute increase in body weight were comparable in normal mice and AbIA mice during the experiment.

Representative histopathology of the front paw of AbIA mice. (A) Normal mouse control; (B) vehicle-PLGA nanoparticle treated AbIA mouse; (C) PLGA-nanosteroid (30 μg ×1×, IV) treated AbIA mouse. An arrow indicates cellular infiltrations. A yellow bar indicates 5 mm.
The arthritic mice treated with saline or vehicle-PLGA showed extensive cellular infiltration (3.9 (0.9)), although cartilage erosion was negligible even in the mice with severe arthritis. In contrast, PLGA-nanosteroid treated mice showed only mild cellular infiltration (0.9 (0.7)) (p< 0.01 v vehicle control) (fig 5⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histology score of AbIA mice. The extent of cellular infiltration in AbIA mice treated with PLGA-nanosteroid (30 μg ×1, IV), BSP (30 μg, ×3, IV), or vehicle-PLGA nanoparticles was determined and graded from 0 to 3 in each paw. Data represent mean (SEM) (seven mice in each group). *p<0.05 (PLGA-nanosteroid v BSP); †p<0.01 (PLGA-nanosteroid v vehicle-PLGA nanoparticles).
DISCUSSION
The results indicate that PLGA encapsulation with this new method can strongly increase the therapeutic efficacy of BSP as an intravenous treatment for inflammatory diseases, including RA. Preparation of the PLGA nanoparticles encapsulating a water soluble bioactive molecule such as BSP and designed for sustained release and intravenous injection has been described previously (J Controlled Release, in press). Hydrophilic BSP is converted to a hydrophobic form by interaction with a zinc ion and was encapsulated in the nanoparticles of PLGA with a size of 100-200 nm. The resulting nanoparticles, ranging from 90 to 120 nm in diameter with a BSP content of 2–3 weight per cent, were used in this study. BSP was gradually released from the nanoparticles in the diluted serum in vitro over 20 days without any apparent initial burst (J Controlled Release, in press). More than 95% of the PLGA nanoparticles with rhodamine was trapped in the spleen and the liver, whereas 2–3% of them accumulated in the inflamed, but not in the intact, joint (data not shown). The PLGA-nanosteroid incorporated in the spleen and the liver did not exhibit anti-inflammatory effect, because we could not show any detectable amounts of free BSP in the serum after the PLGA-nanosteroid treatment, and this treatment affected neither the delayed-type hypersensitivity nor the T cell proliferation in rats (data not shown).
Nanoparticles can target a specific lesion and are less likely to burst at an early stage of administration, therefore enabling them to gradually release the agent at the site of the lesion over a prolonged period. It has been demonstrated that the nanoparticles encapsulating betamethasone dipropionate, a hydrophobic steroid, released a significant fraction of betamethasone at an early stage, with about 90% or more of betamethasone having been released after several days. However, the nanoparticles prepared according to the method of this study, in which the initial bursting out of the steroid is significantly reduced, released the steroid more gradually and over an extended period of time. A targeted intravenous drug delivery approach using the PLGA-nanosteroid could be a highly attractive alternative to current glucocorticoid treatment strategies such as intravenous pulse therapy or local intra-articular injections. We could reduce the number of steroid administrations using PLGA, whose safety has been clinically proved.10–13,25 Although the effectiveness of liposome-steroid for the treatment of arthritis has been described,8 it is difficult to maintain the prolonged release by liposome preparation. PLGA-nanosteroids provide an opportunity to achieve high concentrations of glucocorticoid delivered selectively to all inflamed arthritic joints by a simple intravenous injection.
In a preliminary study in AA rats, different sizes (70, 100, and 190 nm) and different surfactants (lecithin, polyvinyl alcohol, and Pluronic F68) of the PLGA nanosteroid have produced the same anti-inflammatory effects. Moreover, no significantly different anti-inflammatory effects between PLGA (mol wt 13 000), PLA (mol wt 14 000), and PLGA (mol wt 8000) nanosteroids were found. It has also been demonstrated that nanoparticles of PLGA or PLA with small molecular weights, tend to release the steroid at an earlier stage, and that the nanoparticles of PLGA tend to release the steroid earlier than those of PLA. Furthermore, selective accumulation of 100–200 nm, but not 500 nm, PLGA nanoparticles at an inflammation site has been shown in rats using fluorescence labelling experiments (data not shown). Hepatosplenic uptake is largely responsible for the elimination of 500 nm nanoparticles from the circulation and appears to be mediated by residual macrophages in these organs. We then used PLGA (mol wt 13 000) nanosteroid of 90–120 nm in size with lecithin as a surfactant in these experiments. A single intravenous injection of the nanosteroid can induce a strong, rapid, and longlasting therapeutic benefit. About 30% remission of joint inflammation can be accomplished within 1 day of treatment and the therapeutic benefit of the injection lasts for up to 1 week, whereas the equivalent dose of limethasone (lipid emulsion; dexamethasone palmitate), which is currently employed for the treatment of RA,26–,28 gradually loses its anti-inflammatory effect over time (data not shown). Earlier treatment with the PLGA-nanosteroid might also prevent cartilage erosion in AA rats.
In AbIA mice, the same prolonged anti-inflammatory effect could also be obtained with the PLGA-nanosteroid within 1 day and lasted for 7 days. The anti-inflammatory effect might be equivalent to polyethylene glycol-liposomal glucocorticoid as described before.8 PLGA nanoparticles may protect BSP against conversion and degradation in the circulation, as is seen in the liposome. It may also prevent it from the rapid and extensive tissue distribution that occurs with free BSP. PLGA nanoparticles may not only enhance the concentration of the drug at the target site but also lower drug concentration at non-target tissues. The possibility of reduced toxicity may further improve the therapeutic index of the glucocorticoid upon PLGA nanoparticles.
In conclusion, the results of this study indicate that a single intravenous dose of hydrophilic BSP encapsulated in PLGA nanoparticles can lead to a rapid, complete, and durable resolution of joint inflammation owing to the enhanced and preferential localisation of BSP in the inflamed joints. This new approach may offer important advantages over existing treatments in arthritis, such as pulse therapy, intra-articular injection, and liposome preparations of steroids. Additionally, these results might apply to other inflammatory or immune diseases for which a steroid is effective. We have also observed the effectiveness of this preparation in autoimmune uveitis models in rats (in preparation). As compared with free BSP, the slow release, PLGA based BSP nanoparticles are safe and markedly reduce the clinical score of experimental arthritis in rodents.
Acknowledgments
We thank Ms S Asano and Ms L Yazaki for their superb technical assistance.
This work was supported in part by a Health and Labour Science Research Grant from the Health and Labour Ministry of Japan (H14- Nano017).
REFERENCES
Footnotes
Published Online First 4 February 2005