
Abstract
Darifenacin hydrobromide is a selective muscarinic M3 receptor antagonist that is indicated for use in treatment of overactive bladder disorder. Darifenacin was found to have a short terminal elimination half-life after intravenous and immediate-release oral dosage forms (3–4 hours) but this increased with a prolonged-release (PR) formulation (14–16 hours). The absolute bioavailability of darifenacin from 7.5 and 15mg PR tablets was estimated to be 15.4% and 18.6%, respectively. With repeated once-daily oral administration of the PR formulation, peak plasma concentrations of darifenacin are achieved approximately 7 hours post-dose. After oral administration, darifenacin is well absorbed from the gastrointestinal tract and very little unchanged drug (<2%) is recovered in the faeces. Steady state is achieved after 6 days of once-daily administration of the PR formulation. As expected, values of peak plasma concentration (Cmax) and area under the plasma concentration-time curve are dose dependent, although the increase in plasma concentrations is proportionally greater than the increase in dose owing to saturation of presystemic metabolism. From intravenous administration, it has been established that darifenacin possesses a moderate-to-high hepatic extraction ratio, with high plasma clearance (36–52 L/h) and a volume of distribution (165–276L) that exceeds total body water. It is highly protein bound (98%), primarily to α1-acid glycoprotein. Darifenacin is subject to extensive hepatic metabolism, with 3% of unchanged drug excreted in urine and faeces. Metabolism is mediated by hepatic cytochrome P450 2D6 and 3A4, the main metabolic routes being monohydroxylation in the dihydrobenzfuran ring, dihydrobenzfuran ring opening, and N-dealkylation of the pyrrolidine nitrogen. Several possibly important drug-drug interactions have been identified with darifenacin, including ketoconazole, erythromycin and fluconazole, each of which increases darifenacin mean Cmax by 9.52-, 2.28- and 1.88-fold, respectively. When given with imipramine, darifenacin causes 1.6-fold higher plasma concentrations of the antidepressant and its major metabolite. Moderate hepatic impairment, but not renal insufficiency, has been shown to increase plasma concentrations of the drug. The pharmacokinetic profile of darifenacin is not affected by food.
Similar content being viewed by others
Overactive bladder (OAB) disorder is a common ailment among the general population, affecting women and men of all ages. Symptoms of overactive bladder include urgency, urge urinary incontinence, frequency and nocturia.[1,2] One of the major pharmacological targets in the alleviation of these symptoms is modulation of the cholinergic nervous system, and specifically antagonism of muscarinic activity on the bladder. There are five distinct subtypes of muscarinic receptor, denoted M1 to M5, differing in localisation patterns, including smooth muscle cells, glandular acinar cells, cardiac myocytes and neurons. Functionally, the M1, M2 and M3 receptor subtypes have been well characterised both in vitro and in vivo.[3] Each subtype has tissue-specific physiological functions in the presence of acetylcholine. Of relevance to OAB is the M3 receptor subtype, for which acetylcholine causes smooth muscle contraction of tissues such as gastrointestinal mucosa or detrusor muscle of the bladder.[2,3] The mainstay of treatment for OAB is a combination of behavioural therapy (bladder training) and pharmacotherapy. Currently the most frequently used drug treatment involves antimuscarinic agents, such as oxybutynin and tolterodine.[2,4] These agents are widely used for the symptomatic treatment of OAB, but are not selective for M3 receptors, the subtype thought to be primarily responsible for overactivity of the detrusor muscle of the bladder.[5]
Darifenacin hydrobromide ([(S)-[-2-[1-[2-(2,3-dihydrobenzofuran-5-yl)ethyl]-3-pyrrolidinyl]-2,2-diphenylacetamide; figure 1) is a muscarinic receptor antagonist with selective affinity for the M3 cholinergic class of receptors.[6] As its chemical name denotes, darifenacin contains a centre of asymmetry, but was developed as the single S-enantiomer, which is the eutomer of the racemic compound. In 2004, darifenacin was granted regulatory approval by US FDA and European regulators for use in the US and all 25 countries of the EU, Norway and Iceland for the treatment of OAB.[2,7] More recently, approval has also been granted for Indonesia and Australia. Based on the safety and efficacy results of clinical pharmacology studies, the recommended starting dose is 7.5mg once daily as a prolonged-release (PR) formulation for all patients, but for patients requiring greater symptom relief the dose may be increased to 15mg once daily based on individual response and tolerability.

Structure of darifenacin and its major metabolites, together with its metabolic pathways in humans and recovery, expressed as percentage of total recovery from urine and faeces. EM = extensive metaboliser.
The purpose of this review is to describe the current state of knowledge of the pharmacokinetic properties of darifenacin. This includes data both from the marketed PR formulation and from immediate-release (IR) dosage forms which were the subject of early studies with darifenacin.
1. Analytical Methods
In biological fluids (plasma and urine), concentrations of darifenacin and some of its major metabolites (UK-148,993, UK-88,862 and UK-73,689) have been quantified by use of validated reversed-phase high-performance liquid chromatography (HPLC) methods. For most of the methods used during development of darifenacin, solid-phase extraction was followed by reversed-phase HPLC with subsequent detection of the analytes using atmospheric pressure chemical ionisation liquid chromatography-tandem mass spectrometry (LC-MS/MS).[8] In some variants of the assay procedure, semi-automated methods employing 96-well microtitre solid-phase extraction plates were used for sample clean-up.[8] The validated lower limits of quantitation of these methods were as low as 0.025 ng/mL for darifenacin and 0.05 ng/mL for the metabolite UK-148,993 (figure 1).[8]
2. Pharmacokinetic Properties of Darifenacin
2.1 Absorption Following Oral Administration
Darifenacin is rapidly and completely (∼97%) absorbed following oral administration, as seen by findings of very little unchanged darifenacin recovered in faeces in human extraction studies.[9,10] The rate of absorption of darifenacin from PR tablets is limited by the rate of drug release from the tablet matrix, resulting in later absorption, taking place mainly from the colon rather than upper gastrointestinal (GI) tract. Thus, administration of darifenacin IR tablets to young, healthy, mostly Caucasian volunteers generally resulted in time to peak plasma concentration (tmax) within 0.5–1.5 hours of tablet administration (table I; figure 2),[9,11–20] compared with 5.5–11.5 hours from PR tablets (table I; figure 2),[13,14] and correspondingly higher peak plasma concentration (Cmax) values than observed after administration of PR formulations (table I).[13,14] The rate of absorption from the colon appears to be similar to that from the upper GI tract, as seen by the tmax following administration by intubation directly to the ileocaecal junction compared with that following an oral solution (studies I and II, table I).[11,12] In addition, the terminal elimination half-life (t½β) following oral solution administration (3.1–7.1 hours) closely matched the t½β observed after ceasing intravenous infusion (study I, table I).[11,12,20] In contrast, the t½β after administration of the PR formulation appeared to be considerably longer (6.7–18.8 hours; table I).[19]


After administration of PR formulations, the relative bioavailability of darifenacin is approximately 2-fold higher than that of equivalent doses of IR formulations.[13,14] Circumvention of intestinal first-pass metabolism has been reported to contribute to higher bioavailability of PR formulations of other medications.[21] However, this does not completely account for the difference with darifenacin. This is illustrated by the finding that although colonic administration of 15mg darifenacin increased systemic bioavailability by 51% relative to a 15mg dose of oral solution (study II, table I),[12] this represented an increase from 12% only to 18% of the systemic bioavailability of an intravenous dose (study I, table I).[11] The proportion of dose not crossing the gut wall, calculated on the basis of systemic and hepatic availability (i.e. 27%, derived from the hepatic extraction ratio of 0.73), decreased from 55% from the oral solution to 36% for the colonic administration, but was still substantial. Similar conclusions can be drawn for the PR tablet, given the comparable bioavailability of this formulation and the colonic solution in the same study (table I); in this case, it is estimated that 30% of the 20mg PR dose did not cross the gut wall.
The area under the plasma concentration-time curve (AUC) of darifenacin was observed to increase with dose (table I), although at higher dose levels the increase in AUC appears to be disproportionate. By pooling the available data from 20 phase I studies involving repeated-dose administration of darifenacin, a nonlinear relationship was apparent in the daily dose versus AUC relationship where the relative increase in AUC exceeded the increase in dose level. This was also established in a population pharmacokinetic analysis involving phase I and phase II studies.[22] Because no such nonlinearity was observed after intravenous infusion (table I, study 1),[11] saturation of clearance (CL) cannot explain the finding after oral doses. The population analysis identified a relationship between dose and bioavailability, wherein bioavailability was observed to increase as the oral dose increased, presumably as a consequence of saturation of presystemic metabolic activities. Although the precise mechanisms have not been elucidated, darifenacin is a known substrate of cytochrome P450 (CYP) pathways (see section 2.3.1), and therefore this saturation may potentially involve intestinal CYP3A4 activities. The absolute bioavailability of darifenacin from 7.5 and 15mg PR tablets at steady state is estimated to be 15.4% and 18.6%, respectively, in subjects who are extensive metabolisers (EMs) via CYP2D6 (see section 3.3).
In vitro studies involving human intestinal CaCo-2 cell monolayers have been conducted to better understand the absorption process of darifenacin. Darifenacin (25 µmol/L) fluxed in the apical to basolateral direction with an apparent permeability of 19 × 10-6 cm/sec, and in the basolateral to apical direction with an apparent permeability of 22 × 10-6 cm/sec.[23] These results indicated that darifenacin readily crosses cell membranes in a non-selective, bidirectional manner. Although darifenacin is a substrate of P-glycoprotein (P-gp) [see section 2.3], which is known to limit the intestinal absorption of some drugs, this appears to apply mainly to moderately permeable compounds[24] and the low levels of unchanged drug excreted in faeces suggest that this mechanism plays little part, if any, in limiting the absorption of darifenacin in vivo.
2.1.1 Effect of Food
In healthy Western (mostly Caucasian) young male volunteers given a single darifenacin 30mg PR tablet while fasted or postprandial, coadministration of the PR tablet with a high-fat meal caused only a mean increase in Cmax of 22%, and a mean reduction in tmax of 3.3 hours (study VI, table I).[16] However, there was no clinically significant increase in the AUC when the drug was given under these two conditions. With the same dosage given daily, no differences were apparent in mean tmax, Cmax or AUC between the fasted and postprandial high-fat meal states (study VII, table I).[17,18] Although total exposure was also increased by food consumption, the difference between fed and fasted states was relatively small. In none of these studies were increases in adverse effects noted when food was coadministered with darifenacin. The effect of food on steady-state pharmacokinetics of darifenacin has also been evaluated. In healthy male volunteers aged 18–45 years given PR darifenacin 30mg daily for 10 days, food had no effect on steady-state darifenacin pharmacokinetic parameters (study VII, table I): the 90% confidence interval of the difference between treatment means in tmax, Cmax and AUC were all within the range 80–125%.[17,18]
2.2 Distribution
Similar to many other acetylcholine receptor antagonists,[25] darifenacin is lipophilic and has a large volume of distribution (Vd) that exceeds total body water. In healthy volunteers given the drug by intravenous infusion, the Vd was calculated to range from 165 to 276L (table I). Using population analysis,[22] the Vd at steady state (Vss) was estimated to be 163.4L. After short intravenous infusions, multiexponential decline is observed in the plasma concentration versus time curves (figure 3).[11] In human plasma subjected to equilibrium dialysis, 14C-darifenacin was observed to be highly bound (94–98%). The concentrations of darifenacin in the plasma involved in these unpublished studies ranged from 20 to 357 ng/mL.[26–29] The human whole blood : plasma concentration ratio of 14C-darifenacin in whole blood at a concentration of 16.8 ng/mL was found to be 0.68, indicating that binding to cells is modest. Assuming a haematocrit of 0.45, the darifenacin concentration in plasma would be 3.4-fold greater than that in blood cells. In vitro, α1-acid glycoprotein possessed a higher binding affinity to labelled darifenacin than that reported for albumin, when isotonic buffer solutions were dialysed against buffer containing darifenacin (100 and 500 ng/mL) in physiologically relevant concentrations of plasma protein.[28] The metabolite UK-148,993 was less highly bound than darifenacin in human plasma, with mean binding of 87%. Therefore, the unbound concentration of UK-148,993 is estimated to be approximately 6-fold higher than that of darifenacin. This would be expected to contribute to a larger Vd for this metabolite, and depending on its intrinsic rate of elimination, possibly increased CL.

Using unlabelled darifenacin and plasma samples representing Cmax from six healthy volunteers given 5mg of darifenacin as capsules,[9] the unbound fraction was found to be 0.02 after subjecting the plasma samples to equilibrium dialysis. These results were in line with those of the samples described earlier in which plasma was spiked with 14C-darifenacin.[26,27]
Currently no information is available on the distribution of darifenacin in human tissues. In one study involving male rats given 14C-labelled intravenous darifenacin, whole body autoradiography revealed that concentrations of drug were highest in most tissues within 5 minutes of administration of a 4 mg/kg dose.[30,31] Drug-related radioactive material was detected throughout the body tissues. At 5 minutes post-injection, the five tissues that had the highest levels of drug were, in descending order, adrenal, small intestine contents, lung, liver and pituitary gland. At 72 hours after administration, appreciable tissue levels of radioactivity were still present in adrenal, small intestine contents and liver. By 72 hours, relatively high radioactivity levels were present in retina as well. It was thought that the drug bound to highly pigmented tissues to a greater extent than to less well pigmented tissues.[30,31] Given its lipophilicity, some degree of CNS penetration of darifenacin may be expected, potentially resulting in adverse events associated with central antimuscarinic activity, such as cognitive impairment.[32] However, this study, albeit in rats, demonstrated a particularly low level of radioactivity in the brain at all timepoints, falling to unquantifiable levels within 24 hours.[30] The low CNS concentrations of darifenacin were also shown in a further rat study assessing the concentrations of 14C-labelled darifenacin in cerebrospinal fluid (CSF) following a single oral dose of 10 mg/kg. CSF levels were approximately one-tenth of those observed in plasma dialysate, and in both fluids were mostly accounted for by the metabolite UK-73,689, with unchanged darifenacin and the metabolites UK-148,993 and UK-346,06 accounting for only 6%, 6% and 7% of the total radioactivity in the CSF, respectively.[30,33] These findings, coupled with active CNS efflux mediated by P-gp (see section 2.3.3), suggest that darifenacin has a particularly low potential for eliciting CNS adverse effects, an important consideration in the treatment of older patients in particular. Indeed, in a randomised, double-blind, placebo-controlled, crossover study in 129 elderly patients (aged 65–84 years), PR darifenacin 7.5 or 15mg once daily was not significantly different from placebo in effects on cognitive function.[34]
Using equilibrium dialysis, human plasma protein binding of the metabolites UK-88,862 and UK-73,689 at concentrations of 100–200 ng/mL was 34% and 83%, respectively.[29] This was considerably lower than that of the parent drug.
2.3 Elimination
Clearance of darifenacin is primarily achieved through metabolism, with negligible renal clearance (CLR) and <3% of dose excreted unchanged (2% in urine and 1% in faeces). Mean plasma CL of darifenacin after intravenous infusion to healthy volunteers has been estimated to range from approximately 36 to 52L plasma/hour (table I). When corrected for the mean blood : plasma ratio, the corresponding mean value of blood CL is between 53 and 76L blood/hour. An assumed average hepatic blood flow of 90 L/h in conjunction with a large proportion of CL being attributed to the liver confers on darifenacin the pharmacokinetic properties of a moderately to highly extracted drug; the hepatic extraction ratio of darifenacin is estimated to be 0.73.[11] It is estimated that 70% of dose reaches the portal vein (determined as a percentage of systemic availability/hepatic availability, 19/27), suggesting that the contribution of gut wall first-pass extraction mediated by CYP3A4/CYP2D6 is <27% (with ~3% of dose excreted unchanged). Using nonlinear mixed-effects modelling and a population approach, a CL of 40.2L plasma/hour was determined for a typical Caucasian man homozygous for CYP2D6 EM phenotype.[22] The same analysis determined that the active metabolite of darifenacin, UK-148,993, has a much larger CL (188L plasma/hour) and Vd (499L) than the parent drug. These relatively high estimates of CL are in line with the absolute bioavailability estimates of darifenacin, which are less than 20% for both doses of the PR formulation in EM subjects.
2.3.1 Metabolism
The elimination profile of darifenacin under steady-state conditions has been elucidated using mass balance studies, which show that darifenacin is mostly removed from the body by metabolism, primarily by the liver, with the formation of numerous metabolites. Healthy volunteers received darifenacin 5mg three times daily for 6 days, including 14C-darifenacin as the first dose on day 6, after which faeces and urine were collected for 9 days.[9,10] Excretion of 14C was balanced, with 44% of dose excreted in faeces and 58% in urine, and was essentially complete by 216 hours post-dose. Several metabolites were detected in urine and in faeces, and unchanged darifenacin was present only as a minor component.[9,10]
Identification of these metabolites indicates that darifenacin metabolism is facilitated by three primary routes (figure 1).[9] The first pathway involves monohydroxylation, predominantly of the dihydrobenzfuran ring, thus producing UK-148,993. Another important route involves dihydrobenzfuran ring opening. A third major route involves N-dealkylation of the pyrrolidine nitrogen, thus yielding UK-73,689 and UK-88,862. Glycine conjugation of UK-73,689 ensues, while UK-148,993 is subject to further N-dealkylation. The three major circulating metabolites are UK-148,993, UK-88,862 and UK-73,689, of which only UK-148,993 possesses significant antimuscarinic activity.
In vitro studies using microsomes to determine the enzyme pathways involved in metabolism of darifenacin demonstrated that darifenacin is metabolised via CYP2D6 and CYP3A4 pathways.[35–38] Hydroxylation of darifenacin to form UK-148,993 was mediated by high affinity (Michaelis-Menten constant [Km] 4 µmol/L) and low affinity (Km 47 µmol/L) components.[36] Inhibition studies with quinidine and ketoconazole showed that the high-affinity component comprised both CYP2D6 and CYP3A4, whereas the low-affinity component was entirely CYP3A4. Similar studies on the N-dealkylation of darifenacin to UK-88,862 (plus UK-73,689) showed that this route of metabolism was mediated by CYP3A4 only (Km 102 µmol/L).[37] The ring-opening route was not specifically explored using in vitro studies because UK-222,247 is not formed in microsomes, but the chemical structures suggest that this route of metabolism is also likely to be mediated via CYP2D6 and/or CYP3A4.
2.3.2 Circulating Levels of Metabolites
The metabolite UK-148,993 attains plasma concentrations that are slightly in excess of the parent drug. After doses of darifenacin 7.5, 15 and 30mg PR tablets once daily to young, healthy volunteers, mean drug to mean metabolite ratio ranged from 0.76 to 0.94 for both AUC and Cmax (study IV, table I and table II).[14] For these doses, the mean tmax of UK-148,993 was attained between 4.2 and 5.9 hours, which was slightly lower than the corresponding mean tmax for the parent drug (7–8.1 hours). Although concentrations of metabolite are similar to those of the parent drug, because of the higher unbound fraction in plasma, the concentrations of unbound UK-148,993 are actually much higher than those of unbound darifenacin.

Mean UK-148,993 pharmacokinetic parametersa after administration of prolonged-release tablets of darifenacin to healthy volunteersb
Circulating concentrations of darifenacin and its R-enantiomer have also been assessed by chiral LC-MS/MS after solid phase extraction in human plasma of subjects given darifenacin 30mg once daily as PR tablets.[36] Although measurable levels of darifenacin were found in most samples, its antipode was not detected in any sample, thereby establishing that darifenacin does not undergo appreciable chiral inversion in humans.
2.3.3 Excretion
A negligible amount of darifenacin (~3%) is excreted directly unchanged into the urine.[9] However, urine is a major pathway of excretion of the formed metabolites in humans, with approximately 60% of the dose excreted as metabolites in urine and approximately 40% excreted as metabolites in faeces.
In one study,[39] the affinities of darifenacin and UK-148,993 for P-gp were assessed using cell membranes from insect cells expressing cloned human P-gp. Membranes were incubated in a solution containing adenosine triphosphate (ATP) in the presence of increasing concentrations of darifenacin or UK-148,993. ATPase activity was used as a marker of P-gp activity, with ATPase activity being determined from measurement of the rate of inorganic phosphate release from ATP. Darifenacin and UK-148,993 both stimulated P-gp activity in a concentration-dependent manner. Michaelis-Menten saturation kinetics confirmed that both darifenacin and UK-148,993 were P-gp substrates with mean apparent Km values of 54 ± 23 and 424 ± 190 µmol/L, respectively, with darifenacin having greater affinity in vitro for P-gp than its active metabolite. Although intestinal P-gp activity does not appear to limit darifenacin absorption (see section 2.1), as indicated by the low levels of unchanged drug in faeces, the potential role for this enzyme in excretion of the metabolite is not known.
3. Effect of Intrinsic Factors on Darifenacin Pharmacokinetics
3.1 Age
The steady-state pharmacokinetics of darifenacin from PR tablets in older individuals have been assessed in two unpublished studies.[19,40] In the first study, 62 healthy male and female volunteers aged 50–78 years received repeated daily doses of 30 or 60mg for 14 days (study VIII, table I).[19] Plasma concentrations of darifenacin and UK-148,993 were measured before drug administration on days 1 to 14, and the steady-state plasma concentration profile was determined by serial blood sampling after the dose on day 14. In these subjects, darifenacin AUC after the 60mg dose was somewhat higher than expected. Total exposure from the 60mg dose was approximately 200% higher than that from the 30mg dose (AUC from 0 to 24 hours [AUC24] 775 vs 254 ng · h/mL), and there were similar trends in steady-state values of Cmax, average plasma concentration (Cav) and minimum plasma concentration (Cmin). Despite the apparent nonlinearity, darifenacin doses up to 60mg had no effect on heart rate or heart rate variability.[19] At the 60mg dose there was no further improvement in urinary flow parameters but the overall incidence of adverse events increased in a dose-related manner, primarily as a consequence of a higher incidence of episodes of dry mouth and constipation at the 60mg dose.
Although this study was not designed to specifically address age-related effects on darifenacin pharmacokinetics, the mean AUC in these older subjects was somewhat higher than in other studies involving younger male subjects (e.g. for a 30mg dose the mean AUC was 254 ng · h/mL in this study versus 188 ng · h/mL in study V, table I). This finding is supported by the results from the second study, in which the pharmacokinetics of a 10mg single dose of darifenacin were compared for healthy volunteers aged 21–43 years (n = 13 males) and 64–85 years (n = 15, including 8 males), resulting in an AUC ratio in males (young : elderly) of 65.2%.[40]
The effect of age on the pharmacokinetics of darifenacin was further assessed using data from phase I, phase II and phase III studies. The phase III population pharmacokinetic modelling (using data from 1419 plasma samples from 518 subjects in three studies) suggested that there is no effect of age on CL up to the age of 60 years and then only a small reduction in the CL of darifenacin over the age of 60 years (19% for every decade over 60 years).[41] Thus, the standard dosage regimen allowing individualisation of dose (7.5mg increasing to 15mg daily as required) permits a balance between safety and efficacy in all age groups without specific guidance for tailoring dose according to age.
3.2 Race
Several studies have examined the pharmacokinetics of darifenacin in Japanese young, healthy volunteers. Some of these studies were performed early in the development phase of darifenacin and involved comparisons between the IR and PR formulations. For example, in one such study involving Japanese volunteers (table I),[13] IR formulations yielded Cmax values that were 1.7- to 3.5-fold higher than that obtained after administration of PR formulations containing the same amount of darifenacin. More importantly perhaps, the relative bioavailability after administration of equivalent doses of IR and PR formulations was approximately double in the PR formulations (table I).[13] This was in line with the findings in Caucasian subjects, as discussed in section 2.1.
Although there were differences in study design, including dose levels and single- versus multiple-dose administration, there was some suggestion from the studies involving Japanese volunteers that the exposure to darifenacin might be lower in Japanese than in Western subjects (table I). This was confirmed using a population analysis of the data, in which the bioavailability of the drug was estimated to be 55% lower in Japanese than in Caucasian subjects.[9]
Currently, it is not possible to make any conclusions about the possible influence of ethnic background on darifenacin pharmacokinetics among non-White subjects in the Western studies that have been performed. Over 95% of subjects in Western phase I and phase III studies were White and the influence of race on the pharmacokinetics of darifenacin was not specifically investigated in any study.
3.3 Cytochrome P450 (CYP) 2D6 Genotype
A subset of individuals lack CYP2D6 enzyme activity (e.g. ∼5–10% of the Caucasian population)[42] and a number of polymorphisms in the CYP2D6 gene have been proven to affect directly the pharmacokinetics of CYP2D6 substrates in a clinically relevant manner.[43] One route of darifenacin CL is metabolism via the CYP2D6 pathway and hence darifenacin pharmacokinetics may be affected by variations in the activity of this pathway.
Darifenacin exposure at steady state is higher among individuals who have lower CYP2D6 activity; on average poor metabolisers (PMs) have higher peak and total darifenacin exposure than EM subjects (table III).[44] From comparisons of darifenacin exposure among subjects who received repeated doses of darifenacin PR tablets in any phase I study it is estimated that steady-state total exposure was, on average, 164% higher in PM than in EM subjects following administration of 7.5mg PR tablets, and 99% higher following 15mg PR tablets once-daily administration. Corresponding values for mean peak exposure were 145% and 89%, respectively. However, there is considerable intersubject variability in darifenacin exposure in both EM and PM populations; AUC24 coefficient of variation across the dose range tested was 48–71% for EM and 20–61% for PM subjects. As a consequence, there is a high degree of overlap between ranges of individual exposures in EM and PM populations.

Mean steady-state darifenacin exposure from prolonged-release tablets based on pooled data overall and by predicted cytochrome P450 (CYP) 2D6 phenotype[41]
Higher darifenacin exposure in subjects lacking CYP2D6 activity appears to reflect lower apparent oral clearance (CL/F) of darifenacin in these subjects. Although darifenacin exposure, and hence bioavailability, was higher among PM subjects, data did not suggest any differences in darifenacin absorption or distribution. Time to reach steady state (<6 days) does not appear to be affected by CYP2D6 genotype.[44]
The significant influence that CYP2D6 genotype has on darifenacin pharmacokinetics was confirmed by phase I, phase II and phase III population pharmacokinetic analyses.[22,41,45] Genotype was found to be a statistically significant covariate influencing darifenacin bioavailability (all analyses) and CL (phase I only). Within the phase I analysis model CL was 20% lower and bioavailability was 52% higher in PM subjects compared with EM subjects, which corresponded to a longer t½β (3.8 hours in PM vs 3.1 hours in EM) and 92% higher total exposure in PM subjects. Similarly, in the analysis of phase II data, relative bioavailability was 120% higher in PM than EM subjects, and within the models used for analyses of phase III data relative bioavailability was 55% (phase III analysis), 66% (phase III analysis) and 47% (bridging analysis) higher in PM than EM subjects.[41,45]
The proposed standard dosage regimen allowing individualisation of dose (i.e. 7.5mg once daily, increasing to 15mg daily as required) is expected to permit a balance between safety and efficacy to be achieved in both EM and PM populations; no specific guidance is required for tailoring dose according to CYP2D6 genotype.
3.4 Hepatic Impairment
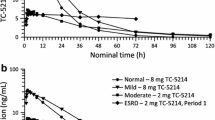
As discussed in section 2.3.1, the CL of darifenacin is primarily achieved through metabolism via CYP3A4 and CYP2D6 pathways. The drug also undergoes extensive presystemic (first-pass) extraction. Hepatic impairment may therefore alter the pharmacokinetic properties of darifenacin. The pharmacokinetics and plasma protein binding of darifenacin have been assessed in subjects with mild and moderate hepatic impairment.[46] The study incorporated a repeated-dose (15mg PR tablets once daily) study design, and steady-state darifenacin plasma concentrations and pharmacokinetic parameters were determined for each category of hepatic function. Patients were classified as being either PMs or EMs of the drug on the basis of their CYP2D6 genotype.
Mild hepatic impairment (Child Pugh A) did not significantly increase the exposure of subjects to darifenacin. Compared with those with normal hepatic function, the patients with mild impairment had a significant increase in CL/F of total darifenacin (table IV).[46,47] Given that in healthy volunteers darifenacin has a high extraction ratio, this finding is perhaps not surprising. The CL of a drug with a high extraction ratio is primarily dependent on blood flow, and in patients with mild hepatic impairment, any decreases in intrinsic CL of unbound drug would not be enough to cause a true change in hepatic CL of the unbound drug. Although the mean was not significantly different, increases in the unbound fraction (table IV) may have occurred in some of the patients with impaired hepatic function, which would have the effect of decreasing the bioavailability of drug by increasing the fraction of the drug eliminated during the first pass through the liver. This apparent decrease in oral bioavailability of darifenacin would result in an increased apparent CL/F in these patients (table IV). Nevertheless, because the unbound AUC and Cmax were not significantly different from those of healthy volunteers, mild hepatic impairment does not seem to significantly alter the pharmacokinetics of darifenacin in a clinically relevant manner.

On the other hand, moderate hepatic impairment (Child Pugh B) had a more pronounced effect on darifenacin pharmacokinetics. Steady-state CL/F of total darifenacin was lower in subjects with moderate hepatic impairment than those with normal hepatic function (56 vs 149 mL/min), and this was reflected in higher peak and total darifenacin exposure in moderately impaired individuals (table IV).[46] The steady-state total darifenacin exposure from PR tablets was significantly (168%) higher in the subjects with moderate hepatic impairment. Correspondingly, steady-state peak darifenacin exposure was 136% higher among subjects with moderate hepatic impairment. There were similar trends in steady-state plasma Cmin and Cav parameters, although overall the peak to trough fluctuation was smaller among subjects with moderate hepatic impairment (table IV). After adjusting for plasma protein binding, steady-state unbound darifenacin exposure was estimated to be 300–370% higher in subjects with moderate hepatic impairment than in those with normal hepatic function. It would appear from these data that under conditions of more severely impaired hepatic dysfunction, the drug was changed from a high to a moderate extraction ratio drug. Based on the changes in unbound AUC and Cmax, adjustments of dosage might be necessary in patients with moderately impaired hepatic function.
The relative effects of hepatic impairment on darifenacin exposure appeared to be broadly similar between EM and PM subjects. Within each category of hepatic impairment darifenacin exposure in PM subjects tended to be higher than that seen in EM subjects (e.g. Child Pugh B mean steady-state total darifenacin AUC24 457 ng · h/mL for PM vs 269 ng · h/mL for EM).[46] The difference in exposure between EM and PM subjects was of similar magnitude when the free drug fraction was taken into consideration. For example, Child Pugh B steady-state unbound darifenacin AUC24 was 13.7 ng · h/mL for PM versus a mean of 10.0 ng · h/mL for EM (n = 1 and n = 5, respectively). In each comparison, darifenacin exposure in PM subjects overlapped the range seen in EM subjects and although darifenacin exposure tended to be higher in PM subjects, exposure in PM subjects with hepatic impairment was not clearly different than in EM subjects with hepatic impairment.
In order to explore whether serum biochemistry parameters are predictive of darifenacin exposure in subjects with hepatic dysfunction, darifenacin AUC24 values were plotted against selected laboratory parameters measured as part of the standard safety monitoring procedures. There were trends indicative of direct relationships between darifenacin exposure and serum bilirubin and alkaline phosphatase concentrations, and inverse relationship between darifenacin exposure and serum albumin concentrations. There was no apparent relationship between darifenacin exposure and ALT, AST or prothrombin time. The apparent relationships between darifenacin exposure and laboratory safety parameters were consistent with data from phase II and phase III studies.
Based on these findings, no specific guidance is required for use of darifenacin in individuals with mild hepatic impairment, but the maximum daily dose of darifenacin should not exceed 7.5mg for patients with moderate hepatic impairment. Darifenacin is not recommended for use in patients with severe hepatic impairment.
3.5 Renal Impairment
Darifenacin is mainly cleared by metabolism (figure 1), and CLR of darifenacin is negligible. For this reason, changes in renal function are unlikely to have a direct effect on darifenacin CL. However, renal disease can cause changes, such as reduction in albumin concentrations, which may affect plasma concentrations or protein binding. Because darifenacin may be prescribed to patients with impaired renal function, darifenacin pharmacokinetics from PR tablets and protein binding were assessed in 23 subjects with varying degrees of renal function ranging from normal function (creatinine clearance [CLCR] >80 mL/min) to severe renal impairment (CLCR <30 mL/min).[46] Steady-state darifenacin pharmacokinetic parameters were determined, and the darifenacin CL/F versus CLCR values were plotted for each subject.
Renal impairment had no clinically relevant effect on the pharmacokinetics of darifenacin. Regression analysis showed that there was no relationship between darifenacin CL and renal function; a regression line (figure 4) fitted to log CL/F and CLCR data had a slope that was not significantly different from zero. As a secondary analysis, pairwise comparisons of darifenacin exposure and CL parameters were also made between categories of renal impairment. Although there were statistically significant differences in darifenacin exposure and CL between subjects with normal renal function and those with moderate renal impairment, there were no differences between normal and mild or severe renal dysfunction groups. In the subjects studied, no clear relationship was observed between level of renal function and the extent of binding to plasma proteins. Although numbers of subjects were small, estimates of unbound fraction (fu) of darifenacin among subjects with moderate or severe renal dysfunction tended to be lower than estimates from matched healthy volunteers (mean fu: normal 2%, mild 2.3%, moderate 1.2%, severe 1.1%). Hence, unbound darifenacin exposure was not increased by the presence of diminished renal function.

Relationship between darifenacin oral clearance and creatinine clearance in subjects with varying degrees of renal function. CL/F = apparent oral clearance; EM = extensive metaboliser; PM = poor metaboliser.
As anticipated, there was a relationship between the severity of renal impairment and the amount of unchanged darifenacin excreted into the urine. Regression analysis showed that CLR was proportional to serum CLCR. A regression line fitted to log CLR versus CLCR data had a slope which was significantly different from zero (slope 1.495, 95% CI 0.888, 2.103, r2 = 0.598). However, because very little darifenacin is excreted unchanged into the urine, this finding is of no clinical relevance. No specific guidance is required for use of darifenacin in patients with renal disease.
3.6 Overactive Bladder Patient Status
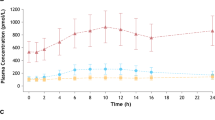
Direct comparison of darifenacin pharmacokinetics between healthy volunteers and OAB patients was not specifically explored, and indirect comparisons were also confounded by differences in age and sex between subjects participating in phase I and phase III studies. Therefore, the similarity of darifenacin pharmacokinetics in healthy volunteers and OAB patients was confirmed by comparison of population pharmacokinetic analyses of phase I and phase III unpublished data.[22,41,45] Although the nature of the data meant that the final structural models differed, both analyses identified dose, sex and CYP2D6 genotype as covariates, and parameter estimates derived from the two analyses were not contradictory. Pharmacokinetic model simulations performed in each analysis for darifenacin PR tablet 7.5, 15 and 30mg daily doses produced similar predictions of total exposure (figure 5). These data suggest that the pharmacokinetics of darifenacin are similar between OAB patients and healthy volunteers.

Comparisons of predicted mean ± SD steady-state area under the plasma concentration-time curve from 0 to 24 hours (AUC24) from population pharmacokinetic analyses of phase I and phase III data. od = once daily.
4. Drug-Drug Interactions
As discussed in section 2.3.1, the in vitro investigations using human liver microsomes have shown that darifenacin is metabolised principally by CYP3A4 and CYP2D6. The metabolism of darifenacin can potentially be inhibited by selective inhibitors of these two enzymes, which may lead to increased circulating concentrations of darifenacin upon coadministration. In addition, darifenacin has shown in vitro potential to inhibit metabolism mediated by CYP3A4 (59–89% inhibition at 100 µmol/L;[48] 50% inhibitory concentration [IC50] 5.3–43 µmol/L for midazolam, testosterone and felodipine[48]) and by CYP2D6 (IC50 1.7–6 µmol/L for bufuralol),[49,50] suggesting that administration of darifenacin may lead to increased concentrations of coadministered substrates of these enzymes.
4.1 Effect of Other Drugs on Darifenacin Pharmacokinetics
4.1.1 CYP3A4 Inhibitors
The effects of various CYP3A4 inhibitors on the in vitro metabolism of darifenacin have been explored.[35–38,48–51] Microsomes from human livers were incubated with darifenacin, in the presence of varying concentrations of ketoconazole, itraconazole, fluconazole, lovastatin, paroxetine or verapamil. Enzyme kinetics were calculated from UK-88,862 and UK-148,993 concentrations in samples taken at intervals. For both UK-88,862 and UK-148,993 metabolic pathways, the most potent inhibition of darifenacin metabolism, was seen with ketoconazole (IC50 0.02 µmol/L for formation of UK-88,862 and 0.01 µmol/L for formation of UK-148,993).[38] A range of inhibitory potencies were observed for the other compounds, with the weakest inhibition being seen for paroxetine and fluconazole.
The effects of ketoconazole on darifenacin pharmacokinetics were confirmed in a crossover study conducted in 16 healthy male volunteers aged 20–52 years (table V).[52,53] Subjects received darifenacin PR tablets 30mg plus either ketoconazole 400mg or placebo once daily for 6 days. Ketoconazole resulted in large increases in darifenacin plasma concentrations. Total AUC was 10.6-fold higher and Cmax was 9.5-fold higher when ketoconazole was coadministered with darifenacin than when darifenacin was taken alone (table V).[52,53] This effect of ketoconazole coadministration was also confirmed by population pharmacokinetic analysis of data generated during development of darifenacin. Using nonlinear mixed-effects modelling, ketoconazole was found to reduce the CL of darifenacin and UK-148,993 by 61% and 68%, respectively, and to increase the bioavailability of darifenacin by 4-fold.[9]

Effect of other drugs on steady-state darifenacin plasma concentrations when coadministered with 30mg darifenacin prolonged-release tablets to healthy male volunteers. Each study involved a crossover placebo-controlled design
Erythromycin was also found to increase darifenacin plasma concentrations in 30 healthy male volunteers aged 19–41 years given repeat doses of darifenacin PR tablets 30mg once daily with and without erythromycin 500mg (table V).[53,56] Coadministration of erythromycin caused 95% and 128% increases in darifenacin AUC and Cmax, respectively. In contrast to ketoconazole, erythromycin was found to have a lesser effect on the plasma concentrations of darifenacin, perhaps because of its 50-fold lower ability to inhibit CYP3A4.[9] Similar results were noted for the moderate CYP3A4 inhibitor, fluconazole (table V).[57] Population pharmacokinetic analysis determined that although erythromycin did not affect darifenacin CL, it did cause an increase of 3- to 4-fold in bioavailability of darifenacin.[9]
Given the results with ketoconazole, the maximum daily dose of darifenacin should not exceed 7.5mg when coadministered with potent CYP3A4 inhibitors (e.g. ketoconazole, itraconazole, miconazole, troleandomycin and nefazadone). No specific guidance is required for use of darifenacin in the presence of moderate CYP3A4 inhibitors, such as erythromycin.
4.1.2 CYP2D6 Inhibitors
Paroxetine is known to be a potent inhibitor of CYP2D6 activity,[58,59] but is not a potent inhibitor of darifenacin metabolism in vitro (IC50 46–74 µmol/L). Furthermore, in vitro studies indicate that inhibition of CYP2D6 has a smaller effect on darifenacin metabolism than inhibition of CYP3A4. When tested in vivo, coadministration of paroxetine 20mg and darifenacin 30mg daily in 26 healthy, young male volunteers caused a small increase in steady-state darifenacin exposure. Darifenacin AUC and Cmax were increased by 33–36% after coadministration of paroxetine (table V).[54] Although this study employed a dose rate higher than that recommended for OAB (15mg once daily), the effect of coadministration of paroxetine with the proposed clinical doses can be predicted. It appeared that the contribution of CYP2D6 to darifenacin metabolism at steady state was inversely related to dose, and is highest at low doses. One consequence is that the difference in darifenacin exposure between EM and PM subjects at steady state would be greater at lower doses. Because coadministration of paroxetine may convert EM subjects to a PM phenotype, by extrapolation, coadministration of a potent CYP2D6 inhibitor with darifenacin is expected to result in a larger percentage change in darifenacin exposure at 7.5 and 15mg daily doses than observed with 30mg.
As expected, coadministration of a CYP2D6 inhibitor had no effect on darifenacin pharmacokinetics in subjects lacking CYP2D6 activity. Although formal statistical analysis was not performed, visual inspection of data indicated that darifenacin exposure was not increased by paroxetine coadministration in PM subjects (darifenacin plus paroxetine group mean AUC24: day 6, 381 ng · h/mL; day 16, 347 ng · h/mL).
Guidance about the use of CYP2D6 inhibitors with darifenacin should match that given for dosage according to CYP2D6 genotype.
4.1.3 Mixed CYP Inhibitors
Because darifenacin is metabolised through both CYP3A4 and CYP2D6 pathways, coadministration of a drug that inhibits both enzymes, such as cimetidine, could potentially cause a different magnitude of effect compared with a drug that inhibits only one pathway. By applying a randomised, crossover design in 20 healthy male volunteers aged 20–32 years, relatively large doses of cimetidine (800mg twice daily) caused changes similar to paroxetine and other less potent CYP3A inhibitors (table V).[55] Coadministration of cimetidine resulted in 34% and 42% increases in darifenacin AUC and Cmax, respectively. Despite the increases, safety assessment revealed that the increase in darifenacin plasma concentrations resulted in minimal increase in incidence of adverse effects.[55]
The change in darifenacin exposure caused by concomitant mixed inhibitor use with 7.5 and 15mg darifenacin doses has not been determined. Although prediction of the magnitude of changes with the proposed clinical doses is problematic, it is anticipated that the percentage changes will be similar across doses.
4.1.4 CYP Inducers
Although there is no evidence to date that CYP2D6 expression can be induced by exogenous agents, a number of drugs are known to induce expression of other CYP enzymes, including CYP3A4. The effect of drug-induced CYP induction on the pharmacokinetics of darifenacin from the proposed PR tablet dosage regimen was assessed from population pharmacokinetic analysis of phase III data (drugs classified as CYP inducers were insulin, nafcillin, omeprazole, carbamazepine, corticosteroids, rifampicin [rifampin], rifabutin, isoniazid, phenobarbital [phenobarbitone], secobarbital [secobarbitone], allobarbital [allobarbitone], methylphenobarbital [methylphenobarbitone], propylbarbital [propylbarbitone], amobarbital [amylobarbitone], butobarbital [butobarbitone], secbutabarbital [secbutabarbitone], pentobarbital [pentolbarbitone], cyclobarbital [cyclobarbitone], phenytoin, primidone and troglitazone).[41] Coadministration of drugs that have the potential to induce CYP expression had little apparent effect on the pharmacokinetics of darifenacin among patients in phase III studies. Of 518 patients included in the initial phase III analysis, 44 received concomitant administration of drugs that have the potential to induce CYP expression. Similarly, out of 829 patients included in a supplementary analysis of phase III data, 60 received agents categorised as CYP inducers. In each case, coadministration of CYP inducers was tested but was not supported as a statistically significant covariate in the final structural pharmacokinetic model, suggesting that the effects of these inducers on darifenacin pharmacokinetics are small in the context of other sources of variability.
At this time no specific dosage recommendations are given for administration of CYP3A4 inducers with darifenacin.
4.2 Effect of Darifenacin on Pharmacokinetics of Other Drugs
4.2.1 CYP3A4 and CYP2D6 Substrates
A series of studies in healthy volunteers have been conducted to assess the effect of darifenacin on the pharmacokinetics of drugs that are substrates of CYP3A4 or CYP2D6 (midazolam, debrisoquine, imipramine).[53,60–63] The CYP2D6 substrate imipramine was chosen as a possible interacting drug with darifenacin because it also has antimuscarinic activity and can be used to treat OAB, and could therefore possibly be taken concomitantly with darifenacin. However, because imipramine is metabolised by CYP2C19, CYP1A2 and CYP3A4 as well as CYP2D6,[64] these other pathways can potentially compensate for inhibition of CYP2D6. To ensure that any effect on CYP2D6 was detected, concentrations of desipramine, a major circulating active metabolite of imipramine formed solely by CYP2D6,[64] were also measured. The effect of darifenacin dosing on CYP2D6 activity in vivo has also been assessed using the debrisoquine : 4-hydroxy-debrisoquine metabolic ratio. Of the CYP3A4 probe substrates tested in vitro the greatest inhibition was observed for midazolam (5.3 µmol/L; with pre-incubation this became 1.2 µmol/L).[48] As a result, midazolam was used as the probe substrate in a clinical study to determine the effect of darifenacin dosing on CYP3A4 activity in vivo.[63] After repeated doses of 7.5, 15 or 30mg daily darifenacin PR were given to healthy volunteers phenotyped to be EM of CYP2D6 in three studies, dose-dependent increases in the debrisoquine : 4-hydroxy-debrisoquine ratio were noted.[53,60–62] After treatment with darifenacin, the ratios increased from approximately 0.6 before treatment to mean values of 2.23, 3.25–4.3 and 7.25 (90% CI 2, 2.7; 2.59, 5.4; and 5.79, 9.06) for the 7.5, 15 and 30mg doses, respectively.[53] This indicated that coadministration of darifenacin inhibited the metabolism of CYP2D6 substrates, but also that the magnitude of the effect depends on the dose of darifenacin used. Notably, use of the proposed clinical doses of darifenacin does not cause complete inhibition of CYP2D6 activity in vivo. The mean debrisoquine metabolic ratio of the EM population remains well below the arbitrary cut-off used to distinguish EM from PM subjects (12.6) even after repeated administration of a 30mg once-daily dose. Furthermore, examination of individual changes in the debrisoquine metabolic ratio indicates that most subjects remain well below the 12.6 cut-off. Inhibition of debrisoquine metabolism declined rapidly after cessation of darifenacin administration and had almost returned to baseline within 3 days of the final darifenacin dose. The offset of CYP2D6 inhibition parallels the anticipated decline in darifenacin and UK-148,993 plasma concentrations, thereby indicating that inhibition is competitive and reversible. After repeat-dose administration of darifenacin PR 30mg to healthy volunteers using a two-period crossover design, it was found that darifenacin caused increases in AUC of imipramine, desipramine and midazolam (table VI).[53,63,65] These studies were conducted using a dose level of 30mg darifenacin, and hence maximised the potential for an interaction to be shown. From the results, the largest observed changes were in the AUC and Cmax of imipramine and its formed metabolite, desipramine. The AUC was observed to increase 0.7-fold (90% CI 0.4, 1) and 2.6-fold (90% CI 1.8, 3.6) for imipramine and desipramine, respectively, after administration of high doses of darifenacin.[53,65]

Effect of darifenacin on steady-state plasma concentrations of other drugs when coadministered with several days of repeated doses of darifenacin 30mg prolonged-release tablets once daily to healthy volunteers. Each study involved a placebo-controlled crossover design
Comparison of these results with published data suggests that the relative potency of CYP2D6 inhibition by darifenacin 30mg once daily is lower than that of the potent CYP2D6 inhibitor paroxetine.[69,70] This finding is consistent with the reported in vitro data where the inhibition constant (ki) for paroxetine against the CYP2D6 substrate bufuralol is 0.8 µmol/L compared with 2 µmol/L for darifenacin. Based on these observations it is therefore believed that darifenacin is not a potent inhibitor of CYP2D6 metabolism.
Administration of darifenacin does not lead to complete inhibition of the CYP2D6 isoenzyme. Nevertheless, caution should be exercised when darifenacin is used concomitantly with medications predominantly metabolised by CYP2D6 and which have a narrow therapeutic window, such as flecainide, thioridazine and tricyclic antidepressants. The increase in midazolam AUC (table VI) after repetitive supratherapeutic doses of darifenacin was significant but relatively modest (17%).[63] Based on this, it appears that darifenacin does not inhibit the CYP3A4 isoenzyme at clinically relevant concentrations. No specific dosage recommendations are given for administration of CYP3A substrates with darifenacin.
4.2.2 Oral Contraceptives
Darifenacin had no effect on the pharmacokinetics of the active components of the Microgynon®Footnote 1 combined oral contraceptive pill (table VI).[66] Pharmacokinetic parameters of levonorgestrel and ethinylestradiol were similar when the pill was administered alone and when coadministered with darifenacin at steady state. Although this study employed three times-daily administration of IR capsules rather than once-daily PR tablets, because the interactions were explored at steady-state darifenacin concentrations using a 30mg total daily dose, the data can be extrapolated to use of the proposed PR tablet dosage regimen. Consequently, coadministration of darifenacin to females using the oral contraceptive should not affect contraceptive effectiveness. No specific dosage recommendations are given for administration of oral contraceptives with darifenacin.
4.2.3 Warfarin
Coadministration of darifenacin 30mg daily with a single 30mg dose of warfarin did not alter the pharmacodynamic effects of warfarin treatment. There were no significant differences in prothrombin time parameters when warfarin was administered in combination with darifenacin compared with warfarin alone (table VI).[68] In addition, no clinically relevant changes in prothrombin time were observed in individual subjects. No specific dosage recommendations are given for administration of warfarin with darifenacin, although measures of warfarin pharmacodynamics such as prothrombin time and international normalised ratio should continue to be monitored.
4.2.4 Digoxin
Coadministration of repeated doses of darifenacin (at steady state) caused a small increase in digoxin exposure (table VI).[67] Digoxin peak concentrations after coadministration of darifenacin were 20% (90% CI 6, 35) higher than when digoxin was taken alone. Similarly, digoxin total exposure was also 16% higher when darifenacin was coadministered (although the 90% CI of the difference between AUC means lay within the range 80–125%) and there was a comparable trend in digoxin trough concentrations. Because digoxin CLR was not reduced when digoxin was given concomitantly with darifenacin, this suggests that the drug-drug interaction is not at the level of digoxin elimination. Given that both darifenacin and digoxin are substrates for P-gp, it is possible that changes in digoxin exposure could be caused by competition between darifenacin and digoxin for P-gp in the lower gut impeding the efflux of digoxin across the intestinal wall (thereby increasing systemic levels). No specific dosage recommendations are given for administration of digoxin with darifenacin.
5. Pharmacodynamic Effects of Darifenacin
Like all other currently available acetylcholine receptor antagonists, darifenacin reversibly binds to varying degrees to each of the five subtypes of human cloned muscarinic receptors. Moreover, comparative studies involving a series of acetylcholine receptor antagonists have indicated that darifenacin has strong affinity for the M3 subtype receptors and a comparative selectivity profile for M3 receptors that is much more favourable than other acetylcholine receptor antagonists. This is a useful property and facilitates the use of darifenacin in the treatment of OAB, providing maximal antagonism of M3 receptors at a given plasma concentration, with 9- to 59-fold less antagonism of other muscarinic subtype receptors.[71] Given these findings, the potential is high for a reduced frequency or severity of untoward anticholinergic effects with darifenacin in the treatment of OAB.
With regard to the impact of this pharmacokinetic and pharmacodynamic profile on clinical performance, pooled analysis has shown darifenacin to be well tolerated,[72] including in patients over 65 years of age.[34,73] The cardiovascular tolerability profile for darifenacin was similar to that of placebo.[72,74] Nervous system, including CNS, tolerability was also similar for darifenacin and placebo[72] or tolterodine 2mg twice daily.[75] This suggests a lack of CNS penetration in accordance with preclinical findings.[30] Preclinical and clinical pharmacodynamic effects are reviewed in greater detail elsewhere.[76,77]
6. Pharmacokinetic/Pharmacodynamic Analysis
With the wide variability observed across the numerous studies conducted with darifenacin (in formulations, doses, populations and study designs), comparison across studies is difficult. Consequently, a combined analysis was conducted using pooled data and a mixed-effects modelling approach. This was initially conducted using phase I data, allowing the construction of an appropriate model to explore the relationship between darifenacin pharmacokinetic parameters and covariates, and establishing the relative potency of darifenacin and its major metabolite on salivary flow.[78] A similar approach was applied to the more sparse darifenacin plasma concentration data from phase II and phase III studies. Using phase III data, the nonlinear mixed-effects model was used to characterise the relationship between darifenacin exposure and efficacy parameters (including the number of incontinence episodes per week, and average volume of urine per voiding) and adverse events (such as dry mouth), and was broadened to incorporate efficacy data from patients for whom plasma concentration data were not available.[41,45]
Assessment of the relationship between the number of incontinence episodes per week and darifenacin dose was conducted using data from 8507 observations from 2339 individuals in six phase II/III studies (table VII). The studies included in this analysis were all multicentre, double-blind, randomised, placebo-controlled, parallel-group studies in adult patients with OAB, collectively assessing a range of doses of darifenacin from 3.75 to 30 mg/day over treatment periods of 6 (one study) or 12 weeks (five studies) [table VII].[79–87] Overall, patients were aged 18–90 years, and approximately 80% of patients were female.

Overview of study populations included in darifenacin pharmacokinetic (PK)/pharmacodynamic analysis. Each study involved a multicentre, double-blind, randomised, placebo-controlled, parallel-group design in patients with overactive bladder. Except study XIII (phase II), all were phase III studies
The number of incontinent episodes per week was modelled as a continuous variable. Factors influencing baseline were identified as weight, sex and race, while age also had an effect among patients older than 60 years. A large placebo effect with a half-life of onset of 10 days was seen in all studies. The drug effect was best described by a maximal effect (Emax) model. From this model it was estimated that the Emax corresponded to a 41% reduction relative to the baseline placebo response and a 6mg dose would produce half of the Emax (ED50). The drug effect was similar across the studies and apparent differences in treatment effects across studies appear to be due to differences in baseline and placebo effect rather than differences in drug response.
To characterise the relationship between average urine volume per voiding and darifenacin dose, 7875 observations were used from 2291 individuals included in the same phase II/III studies (table VII). The final model had three components: baseline, placebo effect and drug effect. The average volume of urine per voiding was estimated to be 156mL at baseline among patients with idiopathic OAB. Placebo treatment produced a small increase in average urine volume per voiding among women (7%) but on average men showed no change from baseline. The placebo response was time dependent (half-life of onset of effect of 18 days) and reached a maximum only after prolonged treatment. The drug effect on average urine volume per voiding was best described by a time-dependent nonlinear Emax model using dose as the independent variable. The model predicted that Emax would be an average increase in volume voided relative to baseline of 49% and that ED50 would be 38mg. For darifenacin doses of 3.75, 7.5, 15 and 30mg, average percentage increases in volume voided over baseline at 12 weeks were 4%, 8%, 14% and 22%, respectively, irrespective of sex, study or diagnosis. Maximum drug response was achieved quickly; the half-life of onset of drug effect was 6 days.
Similarly, micturition frequency was characterised using 8488 observations from 2337 individuals participating in the same phase II/III studies (table VII). The average number of micturitions per day was estimated to be 10.7 at baseline for a typical woman and 11.6 for a typical man. Placebo treatment reduced the average number of micturitions compared with baseline, but the magnitude of the effect differed between studies. At 12 weeks, placebo produced an average 8–15% decrease from baseline. This placebo response was time dependent (half-life of onset of effect of 20 days) and reached a maximum only after prolonged treatment. The drug effect on average number of micturitions per day was best described by a time-dependent nonlinear Emax model using dose as the independent variable. The model predicted that Emax would be an average decrease in the frequency of micturitions relative to baseline of 9% and that ED50 would be 6mg. The predicted effect of drug alone was independent of study and other covariates, and the maximum drug response was achieved quickly (the onset half-life for drug effect was 5 days).
Dry mouth is the most frequently reported adverse effect associated with darifenacin therapy. In a published report, population modelling was used to identify the relationship between salivary flow and plasma concentrations of darifenacin and UK-148,993.[78] Using concentration versus time data from 226 phase I and II subjects (inclusive of data presented in table I), estimates of pharmacokinetic parameters were derived. The concentration data were linked to salivary gland function by introduction of an effect compartment. The maximum effect estimated for darifenacin on reduction in salivary flow was estimated to range from 46% to 59% using the different models tested. After correcting for its larger free fraction in plasma in a receptor-binding model, UK-148,993 was found to have a negligible potency (2.1% that of darifenacin) for reduction of salivary flow.[78]
7. Conclusion
Darifenacin is a novel acetylcholine receptor antagonist that has a relatively high degree of selectivity for M3 receptors, and therefore has potential as a suitable alternative for use in the symptomatic treatment of disorders associated with excessive stimulation of smooth muscle, such as OAB. Early pharmacokinetic and pharmacodynamic studies investigated a range of doses and formulations, and subsequent clinical evaluations led to selection of a PR formulation of 7.5 or 15mg for once-daily administration to maximise clinical efficacy. The pharmacokinetic properties are characterised by large plasma CL (36–52 L/h) and Vd (165–276L). With PR formulation Cmax is reached after approximately 7 hours, t½β is 14–16 hours and steady-state plasma concentration is reached after 6 days. The pharmacokinetic parameters Cmax and AUC are dose dependent, although the increase in plasma concentration is proportionally greater than the increase in dose owing to saturation of presystemic metabolism. Darifenacin is subject to extensive hepatic metabolism by CYP2D6 and 3A4, with 3% of the unchanged drug excreted in urine and faeces, and caution may be required when administering the drug to patients with hepatic insufficiency or when coadministering with potent inhibitors of CYP3A4.
Notes
The use of trade names is for product identification purposes only and does not imply endorsement.
References
Abrams P, Cardozo L, Fall M, et al. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn 2002; 21: 167–78
Hashim H, Abrams P. Drug treatment of overactive bladder: efficacy, cost and quality-of-life considerations. Drugs 2004; 64: 1643–56
Caulfield MP, Birdsall NJ. International Union of Pharmacology: XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 1998; 50: 279–90
Andersson KE. The overactive bladder: pharmacologic basis of drug treatment. Urology 1997; 50: 74–84
Chess Williams R, Chappie CR, Yamanishi T, et al. The minor population of M3-receptors mediate contraction of human detrusor muscle in vitro. J Auton Pharmacol 2001; 21: 243–8
Alabaster VA. Discovery and development of selective M3 antagonists for clinical use. Life Sci 1997; 60: 1053–60
Novartis media releases. Novartis receives European marketing authorization for Emselex® [online]. Available from URL: http://www.novartis.com [Accessed 2005 Feb 27]
Kaye B, Herron WJ, Macrae PV, et al. Rapid, solid phase extraction technique for the high-throughput assay of darifenacin in human plasma. Anal Chem 1996; 68: 1658–60
Beaumont KC, Cussans NJ, Nichols DJ, et al. Pharmacokinetics and metabolism of darifenacin in the mouse, rat, dog and man. Xenobiotica 1998; 28: 63–75
Study report DM11. The excretion and metabolism of [14C]-UK-88,525 in man following administration of multiple oral doses of 5mg three times daily to healthy volunteers. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Study report 137-219. Double blind, placebo controlled, fourway crossover study to investigate the toleration, safety, pharmacokinetics and pharmacodynamics of single escalating doses of intravenous darifenacin in healthy male volunteers. East Hanover (NJ): Novartis, 2001 May 4. (Data on file)
Study report 137-214. An open, randomised, four way crossover pilot study to investigate if site of drug delivery and formulation affects the bioavailability of a single dose of darifenacin. East Hanover (NJ): Novartis, 2002 Mar 21. (Data on file)
Study report JP-95-501. A single blind, placebo controlled, four way crossover, escalating single oral dose study to investigate safety and pharmacokinetics of controlled-release darifenacin and of immediate release darifenacin in healthy male Japanese volunteers. East Hanover (NJ): Novartis, 2002 Sep 23. (Data on file)
Study report 137-218. An open, randomised, four-way crossover study in healthy male volunteers to assess the effect of dose on the pharmacokinetics of darifenacin after controlledrelease matrix tablets (7.5,15 & 30 mg, o.d. for 10 days) and to compare this with immediate release oral capsules (2.5, 5 and 10mg t.i.d. for 10 days). East Hanover (NJ): Novartis, 2002 Mar 27. (Data on file)
Study report JP-95-502. An open, randomised, crossover, single oral dose study to investigate the effect of food on the pharmacokinetics of darifenacin controlled-release tablet in healthy male Japanese volunteers. East Hanover (NJ): Novartis, 2002 Sep 5. (Data on file)
Study report 137-670. The effect of food on the bioavailability of darifenacin controlled-release tablets. East Hanover (NJ): Novartis, 2002 Jan 3. (Data on file)
Nichols DJ. No effects of food on pharmacokinetics of darifenacin in multiple doses [abstract no. 346]. Pharmacol Toxicol 2001; 89 Suppl. 1: 89
Study report 137-226. An open, randomized, two-way crossover, multiple dose study to assess the safety, toleration and pharmacokinetics of darifenacin at steady-state from oral doses (30mg od) of the controlled-release formulation administered to healthy male volunteers in the fed and fasted states. East Hanover (NJ): Novartis, 2000 Jan 18. (Data on file)
Study report A1371015. A randomised, double blind, parallel group placebo controlled study to assess the safety and toleration of multiple oral doses of darifenacin (CR) 60mg once daily. East Hanover (NJ): Novartis, 2002 Feb 8. (Data on file)
Study report 137-221. An open, randomised, three-way crossover study, in healthy male volunteers, to assess the relative and absolute bioavailability of immediate and controlled-release formulations of darifenacin. East Hanover (NJ): Novartis, 2002 Mar 28. (Data on file)
Fuhr U, Staib AH, Harder S, et al. Absorption of ipsapirone along the human gastrointestinal tract. Br J Clin Pharmacol 1994; 38: 83–6
Kerbusch T, Wahlby U, Milligan PA, et al. Population pharmacokinetic modelling of darifenacin and its hydroxylated metabolite using pooled data, incorporating saturable first-pass metabolism, CYP2D6 genotype and formulation-dependent bioavailability. Br J Clin Pharmacol 2003; 56: 639–52
Study report DM26. In vitro permeability of UK-88,525 in CaCo-2 cells. East Hanover (NJ): Novartis, 2003 Apr 9. (Data on file)
Varma MV, Sateesh K, Panchagnula R. Functional role of P-glycoprotein in limiting intestinal absorption of drugs: contribution of passive permeability to P-glycoprotein mediated efflux transport. Mol Pharm 2005; 2(1): 12–21
Brocks DR. Anticholinergic drugs used in Parkinson’s disease: an overlooked class of drugs from a pharmacokinetic perspective. J Pharm Pharm Sci 1999; 2: 39–46
Report DM8. Binding of [14C]-UK-88,525 in the plasma of rabbit, rat, dog and man. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Study report DM8A. Binding of UK-88,525 and UK-148,993 in the plasma of rabbit, rat, dog and man. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Study report DM8B. Protein binding of [14C]-UK-88,525 to human albumin and α-1-acid glycoprotein. East Hanover (NJ): Novartis, 2002 Feb 21. (Data on file)
Study report DM8C. Binding of UK-88,62 and UK-73,689 in human plasma. East Hanover (NJ): Novartis, 2002 Feb 21. (Data on file)
Devineni D, Skerjanec A, Woodworth TG. Low central nervous system (CNS) penetration by darifenacin, a muscarinic M3 selective receptor antagonist, in rats [abstract]. British Pharmacological Society Summer Meeting; 2005 July 6–8; Cambridge
Study report DM7. Tissue distribution of [14C]-UK-88,525 in male and female rats following administration of a single intravenous (4 mg/kg) dose. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Kay GG, Abou-Donia MB, Messer WS, et al. Antimuscarinic drugs for overactive bladder and their potential effects on cognitive function in older patients. J Am Geriatr Soc 2005; 53: 2195–201
Study report DM24. Concentration of [14C]-UK-88,525 in the cerebrospinal fluid of male rats after oral administration (10 mg/kg): relationship to unbound concentration of [14C]-UK-88,525 in plasma. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Lipton RB, Kolodner K, Wesnes K. Assessment of cognitive function in the elderly population: effects of darifenacin. J Urol 2004; 173: 493–8
Study report DM1. In vitro metabolism of UK-88,525. East Hanover (NJ): Novartis, 2001 Dec 21. (Data on file)
Study report DM1A. In vitro metabolism of UK-88,525 in human liver microsomes; enzymology of UK-148,993 formation. East Hanover (NJ): Novartis, 2001 Dec 21. (Data on file)
Study report DM1B. In vitro metabolism of UK-88,525 in human liver microsomes; enzymology of UK-88,862 formation. East Hanover (NJ): Novartis, 2001 Dec 21. (Data on file)
Study report DM1D. UK-88,525 in vitro interaction studies with cytochrome P450 inhibitors and substrates. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Skerjanec A, Devineni D. Affinity of darifenacin for the P-glycoprotein efflux pump: a mechanism contributing to the CNS sparing profile? Newcastle: British Pharmacological Society, University of Newcastle; 2004 Dec 14–16
Study report 137-203. An open, randomised, three way crossover study to compare the pharmacokinetics of darifenacin when given as a solution in the fasted state, capsules in the fasted state and capsules in the fed state. East Hanover (NJ): Novartis, 2001 Sep 4. (Data on file)
Study report phase3. A population pharmacokinetic and pharmacodynamic analysis of darifenacin phase 3 data. East Hanover (NJ): Novartis, 2002 Sept 3. (Data on file)
Xie HG, Kim RB, Wood AJ, et al. Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol 2001; 41: 815–50
Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999; 20: 342–9
NDA21-513, common technical document. Darifenacin summary of clinical pharmacology: module 2.7.2, sections 3.1.2.1 and 3.1.4.4, tables 13 and 23. East Hanover (NJ): Novartis, 2002 Nov 26 (Data on file)
Study report bridging: a population pharmacokinetic bridging analysis of darifenacin. East Hanover (NJ): Novartis. (Data on file)
Devineni D, Skerjanec A, Greig G, et al. Pharmacokinetics of darifenacin, an M3 selective receptor antagonist: effects of renal or hepatic impairment. Proceedings of the British Pharmacological Society [online]. Available from URL: http://www.pa2online.org/Vol2Issue4abst157P.html [Accessed 2006 Feb 27]
Study report A1371005. An open, two period, parallel group study to compare the single and multiple dose pharmacokinetics, safety and toleration of darifenacin patients with chronic stable hepatic disease to healthy subjects with normal hepatic function. East Hanover (NJ): Novartis, 2002 Oct 8. (Data on file)
Study report DM1E. Inhibition of CYP3A4 activity in human liver microsomes by UK-88,525 and metabolites: a comparison with competitor agents. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Study report DM1C. In vitro inhibition studies of UK-88,525 in human liver microsomes. East Hanover (NJ): Novartis, 2002 Jan 23. (Data on file)
Study report DM1G. Inhibition of CYP2D6 metabolism by UK-88,525: effect of pre-incubation. East Hanover (NJ): Novartis, 2001 Oct 22. (Data on file)
Study report DM1H. Inhibition of CYP2D6 activity in human liver microsomes by UK-73,689, UK88,862 and UK-148,993. East Hanover (NJ): Novartis, 2002 Mar 4. (Data on file)
Study report A1371007. A randomised, placebo controlled, two-period cross-over study to investigate the effects of ketoconazole on the pharmacokinetics, pharmacodynamics, safety and toleration of darifenacin at steady state. East Hanover (NJ). Novartis, 2002 Jan 3. (Data on file)
Devineni D, Skerjanec A, Woodworth TG. Drug-drug interaction potential of darifenacin, a muscarinic M3 selective receptor antagonist for the treatment of overactive bladder, with cytochrome P450 (CYP)3A4 inhibitors and substrates [abstract]. AAPS J 2005; 7 Suppl. 2: M1302
Study report A1371008. A randomised placebo controlled, parallel group study to investigate the effects of paroxetine on the pharmacokinetics, pharmacodynamics, safety and toleration of darifenacin at steady state. East Hanover (NJ): Novartis, 2001 Dec 20. (Data on file)
Study report A1371010. A randomised, placebo controlled, two-period cross-over study to investigate the effects of cimetidine on the pharmacokinetics, pharmacodynamics, safety and toleration of darifenacin at steady state. East Hanover (NJ): Novartis, 2002 Jan 18. (Data on file)
Study report A1371022. A randomised, placebo controlled, parallel group study to investigate the effects of erythromycin on the pharmacokinetics, pharmacodynamics, safety and toleration of darifenacin at steady state. East Hanover (NJ): Novartis, 2001 Dec 18. (Data on file)
Study report A1371024. A randomised, placebo controlled, two-period cross-over study to investigate the effects of fluconazole on the pharmacokinetics, pharmacodynamics, safety and toleration of darifenacin at steady state. East Hanover (NJ): Novartis, 2001 Nov 19. (Data on file)
Jeppesen U, Gram LF, Vistisen K, et al. Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol 1996; 51: 73–8
Crewe HK, Lennard MS, Tucker GT, et al. The effect of selective serotonin re-uptake inhibitors on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin Pharmacol 1992; 34: 262–5
Study report A1371023. An open, randomised, multiple dose, two way crossover, two cohort study to demonstrate the bioequivalence of darifenacin when administered as the proposed commercial 7.5 and 15mg tablets and as the 7.5 and 15mg clinical research tablets in the fasted state. East Hanover (NJ): Novartis, 2001 Nov 16. (Data on file)
Study report A1371038. An open study to investigate the effects of multiple doses of darifenacin 30mg controlled-release tablets on the debrisoquine metabolic ratio of healthy male subjects. East Hanover (NJ). Novartis, 2002 Jul 19. (Data on file)
Study report A1371045. An open, randomised, multiple dose two way crossover study to demonstrate the bioequivalence of darifenacin 15mg controlled-release tablets manufactured at the Brooklyn and Barceloneta sites and to investigate darifenacin’s inhibition of debrisoquine metabolism. East Hanover (NJ): Novartis, 2002 Oct 15. (Data on file)
Study report A1371031. A randomised, double blind, placebo controlled, two period crossover study to investigate the effects of multiple dose darifenacin (CR) on the pharmacokinetics of a single oral dose of oral midazolam. East Hanover (NJ): Novartis, 2002 Jan 21. (Data on file)
Dahl ML, Iselius L, Alm C, et al. Polymorphic 2-hydroxylation of desipramine: a population and family study. Eur J Clin Pharmacol 1993; 44: 445–50
Study report A1371009. A randomised, placebo controlled, two-period cross-over study to investigate the effects of darifenacin on the pharmacokinetics, pharmacodynamics, safety and toleration of imipramine at steady state. East Hanover (NJ): Novartis, 2001 Dec 19. (Data on file)
Study report A1371025. An open, randomized, multiple dose study in healthy adult male volunteers to investigate the dose strength equivalence of the darifenacin proposed commercial controlled-release tablets (7.5 mg, 15 mg, and 30 mg), and to investigate the pharmacokinetics. East Hanover (NJ): Novartis, 2002 Sep 5. (Data on file)
Study report A1371021. A double blind, randomised, placebo controlled, parallel group study to investigate the effect of orally administered darifenacin (cr) on the steady state pharmacokinetics of digoxin in healthy male volunteers. East Hanover (NJ): Novartis, 2002 Feb 8. (Data on file)
Study report A1371030. A double blind, randomised, placebo controlled, two-way crossover study to investigate the effect of orally administered darifenacin (cr) on the pharmacodynamics of warfarin in healthy male volunteers. East Hanover (NJ): Novartis, 2002 Mar 2. (Data on file)
Leucht S, Hackl HJ, Steimer W, et al. Effect of adjunctive paroxetine on serum levels and side-effects of tricyclic antidepressants in depressive inpatients. Psychopharmacology (Berl) 2000; 147: 378–83
Albers LJ, Reist C, Helmeste D, et al. Paroxetine shifts imipramine metabolism. Psychiatry Res 1996; 59: 189–96
Wallis RM, Napier CM. Muscarinic antagonists in development for disorders of smooth muscle function. Life Sci 1999; 64: 395–401
Chappie CR, Steers W, Norton P, et al. A pooled analysis of three phase III studies to investigate the efficacy, tolerability and safety of darifenacin, a muscarinic M3 selective antagonist, in the treatment of overactive bladder: a pooled analysis of phase III studies. BJU Int 2005; 95: 993–1001
Foote JE. Treatment of overactive bladder in the older patient: pooled analysis of three phase III studies of darifenacin, an M3 selective receptor antagonist. Eur Urol. Epub 2005 Jun 27
Serra DB, Affrime MG, Bedigian MP, et al. QT and QTc interval with standard and supratherapeutic doses of darifenacin, a muscarinic M3 selective receptor antagonist for the treatment of overactive bladder. J Clin Pharmacol 2005; 45(9): 1038–47
Foote JE, Glavind K, Kay G. Central nervous system (CNS)-related adverse events in patients with overactive bladder (OAB) treated with darifenacin versus tolterodine [abstract]. BJU Int 2004; 94 Suppl. 2: 1134
Haab F. Darifenacin in the treatment of overactive bladder. Drugs Today (Barc) 2005 Jul; 41(7): 441–52
European Medicines Agency (EMEA). Emselex European Public Assessment Report (EPAR): scientific discussion [online]. Available from URL: http://www.emea.eu.int/humandocs/Humans/EPAR/emselex/emselex.htm [Accessed 2005 Feb 13]
Kerbusch T, Milligan PA, Karlsson MO. Assessment of the relative in vivo potency of the hydroxylated metabolite of darifenacin in its ability to decrease salivary flow using pooled population pharmacokinetic-pharmacodynamic data. Br J Clin Pharmacol 2004; 57: 170–80
Study report A1371001. A phase 3, multicenter, double blind, randomized, placebo controlled, parallel group study of the efficacy and safety of controlled-release darifenacin versus tolterodine in the treatment of subjects with overactive bladder. East Hanover (NJ): Novartis, 2002 Aug 6. (Data on file)
Romanzi LJ, Delconte III A, Kralidis G. Impact of darifenacin compared with tolterodine on incontinence episodes in patients with overactive bladder [abstract no. A3522]. Obstet Gynecol 2005; 105: 88S
Study report A1371002. A phase 3 multicentre, double blind, randomised, placebo controlled, dose response study of darifenacin in patients with overactive bladder (urge urinary incontinence). East Hanover (NJ): Novartis, 2002 Feb 18. (Data on file)
Hill S, Khullar V, Wyndaele J-J, et al. Dose response with darifenacin, a novel once-daily M3 selective receptor antagonist for the treatment of overactive bladder: results of a fixed dose study. Int Urogynecol J Pelvic Floor Dysfunct. Epub 1995 Jul 6
Study report A1371011. A phase 3, multicentre, double blind, randomised, placebo controlled, parallel group study of darifenacin versus tolterodine in patients with overactive bladder (urge urinary incontinence). East Hanover (NJ): Novartis, 2002 Apr 15. (Data on file)
Study report A1371012. A phase 2 multicentre, placebo-controlled, double-blind, dose response study of controlled-release darifenacin in patients with overactive bladder (urinary urge incontinence). East Hanover (NJ): Novartis, 2002 Oct 25. (Data on file)
Study report A1371013. A phase 3 multicentre, double blind, randomised, placebo controlled, parallel group study of darifenacin in patients with overactive bladder (urge urinary incontinence). East Hanover (NJ): Novartis, 2002 Apr 15. (Data on file)
Study report A1371041. A phase 3b multicentre, double blind, randomised, placebo controlled, parallel group study of darifenacin in subjects with overactive bladder (urge urinary incontinence). East Hanover (NJ): Novartis, 2002 Nov 5. (Data on file)
Haab F, Stewart L, Dwyer P. Darifenacin, an M3 selective receptor antagonist, is an effective and well-tolerated oncedaily treatment for overactive bladder. Eur Urol 2004; 45: 420–9
Acknowledgements
The preparation of this manuscript was supported by Novartis Pharmaceuticals and editorial services were provided by ACUMED®. The author is an employee of Novartis Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Skerjanec, A. The Clinical Pharmacokinetics of Darifenacin. Clin Pharmacokinet 45, 325–350 (2006). https://doi.org/10.2165/00003088-200645040-00001
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200645040-00001