
Abstract
Atypical antipsychotics have been linked to a higher risk for glucose intolerance, and consequentially the development of type 2 diabetes mellitus (DM2). We have therefore set out to investigate the acute effects of oral administration of olanzapine and ziprasidone on whole body insulin sensitivity in healthy subjects. Using the standardized hyperinsulinemic euglycemic clamp technique we compared whole body insulin sensitivity of 29 healthy male volunteers after oral intake of either olanzapine 10 mg/day (n=14) or ziprasidone 80 mg/day (n=15) for 10 days. A significant decrease (p<0.001) in whole body insulin sensitivity from 5.7 ml/h/kg (=mean, SM=0.4 ml/h/kg) at baseline to 4.7 ml/h/kg (=mean, SM=0.3 ml/h/kg) after oral intake of olanzapine (10 mg/day) for 10 days was observed. The ziprasidone (80 mg/day) group did not show any significant difference (5.2±0.3 ml/h/kg baseline vs 5.1±0.3 ml/h/kg) after 10 days of oral intake. Our main finding demonstrates that oral administration of olanzapine but not ziprasidone leads to a decrease in whole body insulin sensitivity in response to a hyperinsulinemic euglycemic challenge. Our finding is suggestive that not all atypical antipsychotics cause acute direct effects on glucose disposal and that accurate determination of side effect profile should be performed when choosing an atypical antipsychotic.
Similar content being viewed by others


BACKGROUND
Atypical antipsychotics are at least equivalent to typical antipsychotics in treating the positive symptoms of schizophrenia, and seem to be more effective in treating negative, cognitive, and mood symptoms (Conley et al, 1998; Sanger et al, 1999; Kopelowicz et al, 2000). The efficacy against a broader spectrum of symptoms as well as their lower likelihood of extrapyramidal symptoms have led to an increased use of this class of medication (Kasper et al, 1999, 2001).
Currently, there is an ongoing debate on atypical antipsychotic-induced metabolic changes (American Diabetes Association, 2004b; Boehm et al, 2004; Franciosi et al, 2005; Newcomer, 2005). Whereas the FDA stated that all atypical antipsychotics increase the risk for glucose abnormalities but did not discriminate between drugs within the class of atypical antipsychotics (FDA, 2004; FDA requests that a warning be added to the prescribing information for all atypical antipsychotics regarding the risk of hyperglycemia and diabetes), the Consensus Development Conference on Antipsychotic Drugs and Obesity, and Diabetes agreed on an increased risk for diabetes in patients treated with clozapine and olanzapine. Impairment of insulin action was named as one potential mechanism and a clear need for studies in humans investigating adverse effects of antipsychotic medication on β-cell response was expressed (American Diabetes Association, 2004b).
The association of metabolic dysregulations and psychiatric disorders has been first described as ‘endocrine psychosyndrome’ by Eugen Bleuler in 1944 and published in more detail by his son (Bleuler, 1951). Despite the well-described effects of adiposity on glucose metabolism in the general population (Welch et al, 1990; Karter et al, 1996; Macor et al, 1997; Mayer-Davis et al, 1997), the underlying mechanisms for potential adiposity-independent effects of atypical antipsychotics on insulin sensitivity have not been identified yet. While most studies report an increased risk for the development of metabolic abnormalities like glucose intolerance, insulin-resistance, and consequentially DM2 in rodent (Houseknecht et al, 2005) or dog studies (Ader et al, 2005) as well as in patients with schizophrenia (Hedenmalm et al, 2002; Koller and Doraiswamy, 2002; Newcomer et al, 2002; Sernyak et al, 2002; Wilson et al, 2003; Kane et al, 2004; Simpson et al, 2004; Henderson et al, 2005, 2006, Haupt et al, 2007), patients with schizoaffective disorder (Wilson et al, 2003; Simpson et al, 2004) or patients with mood disorder (Gianfrancesco et al, 2003) treated with atypical antipsychotics, other studies fail to confirm these correlations for healthy subjects (Sowell et al, 2002, 2003) or patients with schizophrenia (Kinon et al, 2001).
This study was designed to examine acute to subacute effects of olanzapine or ziprasidone that are not mediated by the well-known effects of adiposity (Welch et al, 1990; Karter et al, 1996; Macor et al, 1997; Mayer-Davis et al, 1997). Studying these effects in healthy volunteers minimizes the effects of lifestyle that may account for some of the impairments in glucose metabolism seen in schizophrenia patients (Mukherjee et al, 1996; Dixon et al, 2000; Ryan et al, 2003).
Glucose intolerance and insulin resistance have been identified as risk factors for the development of DM2 (type 2 diabetes mellitus) and subsequently for cardiovascular diseases (Unwin et al, 2002). Fasting, post-prandial and post-load glucose are significantly driven by insulin secretion and insulin sensitivity. A variety of techniques to measure these two key parameters exist (Haupt, 2006). Whereas estimation of insulin secretion by frequently sampling oral glucose tolerance tests, fasting glucose, and plasma insulin curves cannot reliably relate β-cell response to acute changes in plasma glucose, intravenous glucose tolerance tests, MINMOD calculations (Bergman, 1989), and hyperinsulinemic as well as euglycemic clamps provide a more accurate measure. One method to avoid the glucose–insulin feedback loop is to control the β-cell stimulus by establishing a steady-state level of plasma insulin and holding plasma glucose concentration at a constant level. This allows for direct quantification of whole body insulin sensitivity and can be done by hyperinsulinemic–euglycemic clamp (DeFronzo et al, 1979).
Olanzapine and ziprasidone were selected for this study because each of them seems to reflect another end of the spectrum of metabolic abnormalities. The degree to which oral administration of olanzapine or ziprasidone induces an acute higher risk for development of insulin resistance cannot yet be definitively ascertained from existing reports. To minimize confounding factors such as comedication, comorbidity, life style, and poorly defined effects of underlying disease per se, it is necessary to study the metabolic effects of these drugs in healthy volunteers. We set out to compare the acute effects on insulin sensitivity of both drugs in male healthy subjects by hyperinsulinemic–euglycemic clamp.
METHODS
Subjects
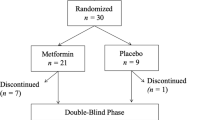
This is an open, randomized, mono-center study of healthy volunteers. We included 30 male subjects between 19 and 41 years of age, with body mass index (BMI) between 18 and 25 kg/m2, fasting glucose level of 100 mg/dl (5.6 mmol/l) or less, normotension, no personal or family history of diabetes, no history of drug or alcohol abuse, no regular nicotine consumption at time of enrolment, and no medication intake for at least 2 weeks before enrolment. None of the volunteers had ever been exposed to any antipsychotic medication.
In each of the volunteers, organic diseases as well as neurologic and psychiatric disorders were ruled out by means of clinical history, physical examination including ECG, hematology, blood chemical study, and a structured clinical interview (Mini-International Neuropsychiatric Interview (MINI); Sheehan et al, 1998) by a rater well experienced in the assessment of this interview before enrolment. The study protocol and all other procedures involved were approved by the local ethics committee of the Medical University of Vienna. After receiving detailed information about all study procedures and possible adverse effects of study medication all subjects had given written informed consent to all procedures before inclusion in the study.
Study Design
After enrolment and completion of the first study day including hyperinsulinemic, euglycemic clamp, and microdialysis, randomization was performed at a 1:1 ratio and subjects were administered for intake of either olanzapine (10 mg/day) at 2000 h or ziprasidone (80 mg/day) at 0800 (40 mg) and 2000 h (40 mg). After 10 days of study medication intake all subjects underwent a second study day including hyperinsulinemic, euglycemic clamp and microdialysis. The last olanzapine dose was taken the night before (10 mg, 2000 h), last ziprasidone dose in the morning of final study day (40 mg, 0800 h), plasma drug-samples were collected during the steady-state clamp to measure concentration of active metabolites (N-desmethylolanzapine/S-methyldihydroziprasidone). Pulse rate (beats/min) and arterial blood pressure (mm Hg) were monitored at 30 min intervals on both study days.
Study Day (days 1 and 12)
After an overnight fast of at least 12 h, the volunteers were admitted to the research ward of the Department of Clinical Pharmacology, Medical University Vienna, and intravenous catheters were placed in the antecubital vein of one arm for infusion of glucose and insulin and in the contralateral wrist vein for blood sampling. Clamp studies were initiated by a primed infusion of insulin until plasma insulin concentration was increased to 100 mU/ml, followed by a constant infusion rate of 1 mU/kg min to maintain this plasma level using a volume-controlled pump (Perfusor, Braun, Germany). KCL was administered at a variable rate necessary to prevent hypokalemia, and 10% glucose was infused at a rate necessary to maintain the blood glucose level between 80 and 100 mg/dl.
Steady-state clamp conditions were reached within 2 h after euglycemic clamp start and maintained at that level for another 2 h. The forearm of each subject was heated by electric pads to arterialize the venous blood (Jansson et al, 1992). Arterialized venous blood samples were drawn every 5 min from the indwelling catheter inserted into a wrist vein to measure the glucose level (Glucotrend®, Roche Diagnostics, Basel, Switzerland). Before the clamp was started, microdialysis probes with a molecular weight cutoff of 20 kDa (CMA-12, CMA Microdialysis, Stockholm, Sweden) were placed in muscle tissue of the thigh. The probes were constantly perfused with Ringer's solution (Mayerhofer, Linz, Austria) at a flow rate of 1.5 μl/min by means of a precision pump (Precidor, Infors-AG, Basel, Switzerland).
Hyperinsulinemic–Euglycemic Clamp
Hyperinsulinemic–euglycemic clamp represents the gold standard methodology (DeFronzo et al, 1979; Haupt, 2006) for the determination of insulin–sensitivity in vivo. This technique measures the amount of glucose required to maintain euglycemia in the setting of an experimentally increased insulin level. While the individual receives a constant insulin infusion i.v. to reach a steady-state insulin level, exogenous glucose is infused simultaneously into a contralateral vein to maintain blood sugar levels at a constant predetermined level (80–100 mg/l). With approximately 70% of glucose disposal coming from skeletal muscle insulin-mediated glucose transport the glucose infusion rate observed provides a reliable measure of whole body insulin sensitivity.
Microdialysis
The principles of microdialysis (MD) have been described previously in detail (Muller et al, 1995; Muller, 2002). In brief, MD is based on sampling of analytes from the extracellular space of tissues by means of a semipermeable membrane at the tip of a MD probe. Microdialysates were analyzed with a Beckman glucose analyzer (Beckman, Brea, CA, USA). A calibration curve was provided by measuring of samples at six different glucose concentrations ranging from 3 to 100 mg/dl. Each sample was measured four times.
Statistical Analysis
One way ANOVA model (one-sided, Bonferroni-corrected) was chosen to analyze the primary outcome measure (whole body insulin sensitivity). Pairwise comparisons within one study group (plasma glucose, plasma insulin and BMI baseline vs endpoint data) were performed using the Wilcoxon matched-pair signed ranks test. Furthermore, an independent sample t-test was performed analyzing drop out rates for both study groups. Level of significance was set at p<0.05. Statistical analyses were performed using SPSS 14.0 (SPSS Inc., Chicago, IL).
RESULTS
Demographics and Adverse Effects
A total of 35 subjects were randomized in the study. Five subjects in the ziprasidone group discontinued after study day 1; four of the five subjects suffered from the following side effects and withdrew consent: one had a panic attack, one developed mild gastroenteritis, one suffered from fatigue and one showed fatigue, vertigo and drowsiness in moderate intensity. One of the five subjects was excluded from the study due to an outbreak of aggression after alcohol intoxication. Thirty subjects completed the protocol, 15 received olanzapine (10 mg/day) and 15 received ziprasidone (80 mg/day). In the ziprasidone group the following mild to moderate adverse events were observed: fatigue in seven subjects, shortened sleep duration in six subjects, in the olanzapine group the most common mild adverse event was fatigue in four subjects. One volunteer had to be excluded from the analysis because he did not reach the expected range of plasma level for olanzapine, 29 subjects entered the data analysis. There was no significant correlation between study-medication and drop out rate.
There was no significant between-study-group difference regarding mean age (olanzapine: 24±3.4 years, ziprasidone: 26±5.7 years; means±SD, Table 1). Plasma levels (Cmax) were in the range of 23±5 ng/ml (mean±SD) for olanzapine and 49±17 ng/ml (mean±SD) for ziprasidone. These plasma levels were reached at 17.86±1.17 h (mean±SD) for olanzapine and 4.4±0.5 h (mean±SD) for ziprasidone (Table 1). Furthermore, there was no significant between-study-group difference in BMI (olanzapine: 22.0±2.2 kg/m2, ziprasidone: 22.6±1.8 kg/m2; means±SM), or mean blood pressure (olanzapine: 83±6 mm Hg, ziprasidone: 83±7 mm Hg; means±SM) at baseline (Table 1).
Subjects in the olanzapine group showed a significant increase (p<0.02) in BMI from 22.0±2.2 (mean±SD) at baseline to 22.6±1.8 (mean±SD) after 10 days of oral intake of 10 mg/day olanzapine. In the olanzapine group, 10 of 14 subjects gained weight over the intake period of 10 days. In the ziprasidone group no significant difference was detected between baseline (22.6±1.8, mean±SD) and endpoint (22.4±1.9, mean±SD) BMI measured after 10 days of oral intake with 80 mg/day ziprasidone (Table 1).
Total Glucose Disposal and Plasma Insulin During the Glucose Clamp
Whole body insulin sensitivity decreased significantly in response to the hyperinsulinemic euglycemic challenge over 120 min (p<0.001). after 10 days of oral intake of 10 mg/day olanzapine (4.7±0.3 ml/h/kg, mean±SM) compared to baseline values (5.7±0.4 ml/h/kg, mean±SM, Figure 1, Table 1). Out of 14 subjects, 13 tolerated less glucose compared to their basal levels. In the ziprasidone group there was no significant change in whole body insulin sensitivity after 10 days of oral intake of 80 mg/day (5.1±0.3 ml/h/kg, mean±SM) compared to baseline (5.2±0.3 ml/h/kg, mean±SM).

Glucose uptake for ziprasidone vs olanzapine. Glucose uptake (rnl/h/kg) during hyperinsulinemic–euglycemic clamp for 120 min comparing baseline values vs values after 10 days of oral intake of either (1) ziprasidone, 80 mg/day, or (2) olanzapine, 10 mg/day in male healthy subjects (means±SM, p<0.001).
The insulin values are shown in Table 2. The insulin values achieved at baseline did not differ significantly between the two groups (6.22±0.78 μU/ml for olanzapine vs 6.29±0.54 μU/ml for ziprasidone, means±SM). The insulin values tended to be higher after intake of olanzapine compared to ziprasidone. After 10 days of oral intake of 10 mg/day olanzapine, significant differences in insulin values were detected at baseline (6.22±0.8 μU/ml vs 9.40±1.0 μU/ml, means±SM, p<0.02), as well as after 60 min (74±4 μU/ml vs 88±6 μU/ml, means±SM, p<0.016), 80 min (71±6 μU/ml vs 92±5 μU/ml, means±SM, p<0.001), and 100 min (78±4 μU/ml vs 98±7 μU/ml, means±SM, p<0.02) of steady state clamp for olanzapine, whereas no significant difference was detected for ziprasidone at any time of the 120 min clamp period. Details are given in Figure 2 and Table 2.

Plasma insulin for ziprasidone vs olanzapine. (Top) Plasma insulin concentrations during hyperinsulinemic–euglycemic clamp (μU/rnl) vs time (min) comparing baseline and after oral intake of ziprasidone, 80 mg/day for 10 days. (Bottom) Plasma insulin concentrations during hyperinsulinemic-euglycemic clamp (μU/rnl) vs time (min) comparing baseline and after oral intake of olanzapine, 10 mg/day for 10 days. These data indicate a significant increase in insulin after oral intake of olanzapine, 10 mg/day for 10 days at baseline (p<0.02), after 60 min (p<0.016), 80 min (p<0.001) and 100 min (p<0.02) of glucose clamp.
After 80 min of hyperinsulinemic euglycemic challenge the glucose uptake rate for the olanzapine group was 6.1±0.4 ml/h/kg (mean±SM) at baseline compared to 4.6±0.2 ml/h/kg (mean±SM) after 10 days of oral intake (10 mg/day) (p<0.015), after 100 min the glucose rate decreased significantly (p<0.001) from 6.5±0.5 ml/h/kg (mean±SM) to 4.6±0.2 ml/h/kg (mean±SM) and after 120 min there was a significant decrease from (p<0.0001) from 6.9±0.5 ml/h/kg (mean±SM) to 4.7±0.2 ml/h/kg (mean±SM). The glucose uptake for the ziprasidone group did not show any significant changes from baseline over time. Details are given in Figure 3.

Glucose uptake for ziprasidone vs olanzapine overtime. (Top) Glucose uptake (ml/h/kg) during hyperinsulinemic–euglycemic clamp vs time (min) comparing baseline and after oral intake of ziprasidone, 80 mg/day for 10 days. (Bottom) Glucose uptake (ml/h/kg) concentrations during hyperinsulinemic–euglycemic clamp vs time (min) comparing baseline and after oral intake of olanzapine, 10 mg/day for 10 days. These data indicate a significant decrease in whole body insulin sensitivity after oral intake of olanzapine, 10 mg/day for 10 days, after 80 min (p<0.015), 100 min (p<0.001), and 120 min (p<0.0001) of glucose clamp.
Glucose Levels Before and During the Glucose Clamp
There was no significant difference in the fasting glucose levels in both groups at baseline (olanzapine: 5.04±0.06 mmol/l, vs ziprasidone: 5.10±0.49 mmol/l, means±SD; Figure 4). In the olanzapine group fasting glucose levels tended to be only mildly higher after the glucose clamp (5.13±0.11 mmol/l vs 4.94±0.11 mmol/l, means±SD) following drug intake of 10 days, but this difference was not statistically significant (Figure 4).

Plasma glucose for ziprasidone vs olanzapine. (Top) Plasma glucose concentrations (mmol/l) during hyperinsulinemic–euglycemic clamp vs time (min) comparing baseline and after oral intake of ziprasidone, 80 mg/day for 10 days. (Bottom) Plasma glucose concentrations (mmol/l) during hyperinsulinemic–euglycemic clamp vs time (min) comparing baseline and after oral intake of olanzapine, 10 mg/day for 10 days.
Regarding the a-i (arterial-interstitial) gradient during the glucose clamp there was an increase in the olanzapine group from 1. 62±1.05 to 3.01±1.37 mmol/l (means±SD) after 10 days of drug intake. For the ziprasidone group a mild decrease from 2.80±0.47 to 2.64±1.44 mmol/l (means±SD) after oral intake for 10 days could be detected. These differences did not reach statistical significance.
DISCUSSION
This study is the first to demonstrate consistent in vivo evidence in humans that olanzapine causes significant acute insulin resistance. In contrast to previous reports (Sowell et al, 2002, 2003) our clamp experiments show that there is a significant decrease (p<0.001) in whole body insulin sensitivity in response to hyperinsulinemic euglycemic challenge in healthy subjects (baseline: 5.7±0.4 ml/h/kg, mean±SM) following oral intake of 10 mg/day olanzapine (4.7±0.3 ml/h/kg, mean±SM, Figure 1). Whereas Sowell et al reported an increased total insulin response for olanzapine, they attributed those changes to the antipsychotic-induced weight gain and concluded that the observed changes were insignificant (Sowell et al, 2002).
We observed several other effects of olanzapine intake on glucose metabolism in healthy male volunteers.
One metabolic effect is a significant increase in BMI (p<0.02) from 22.0±2.2 (mean±SD) at baseline to 22.6±1.8 (mean±SD) following olanzapine administration for 10 days. This finding is consistent with the strong correlation between BMI and insulin resistance from clamps in normal-weight individuals (Lillioja and Bogardus, 1988). A previously proposed hypothesis by Boehm et al (2004) that weight gain represents a risk factor only in the general population but is not associated with the risk of DM2 during antipsychotic treatment does not seem very likely given the evidence for a higher rather than a lower prevalence of DM2 in patients suffering from a major mental disorder in almost all data sets published to date (Mukherjee et al, 1996; Dixon et al, 2000; Okamura et al, 2000; Mokdad et al, 2001; Haupt and Newcomer, 2002; Newcomer et al, 2002; Ryan et al, 2003; Newcomer, 2004; Haupt et al, 2007).
A second metabolic effect is depicted in Figure 2 and Table 2. We found significantly higher plasma insulin values for the olanzapine group after drug intake for 10 days. Significant differences for olanzapine were detected at baseline (6.22±0.8 vs 9.40±1.0 μU/ml, means±SM, p<0.02), as well as after 60 min (74±4 vs 88±6 μU/ml, means±SM, p<0.016), 80 min (71±6 vs 92±5 μU/ml, means±SM, p<0.001), and 100 min (78±4 vs 98±7 μU/ml, means±SM, p<0.02) of steady-state clamp conditions. This parameter also represents a well-studied indicator for insulin resistance (Holmang et al, 1998).
A third metabolic effect observed was the mild increase in arterial plasma glucose obtained during glucose clamp from 4.94±0.11 mmol/l (mean±SD) at baseline compared to levels obtained during glucose clamp after olanzapine intake for 10 days (5.13±0.11 mmol/l, mean±SD). Although these results were not significant, it is noticeable that insulin infusion during clamp induced a gradual increase in the glucose a-i concentration difference from 1.62±1.05 vs 3.01±1.37 mmol/l (means±SD) following 10 days of oral intake of olanzapine, 10 mg/day.
As subjects who were administered with ziprasidone took a 40 mg dose in the morning and 40 mg in the evening, this group experienced more side-effects during the day whereas olanzapine doses were administered only in the evening (due to different plasma half-lives; Miceli et al, 2000; Perry et al, 1997) and felt fatigue mostly during night time when they could sleep. This difference in administration could explain the drop out rate for the ziprasidone group (n=5). We performed a t-test analyzing the drop out rates for both study groups. Even though we could not find a significant difference (p>0.05), this might be due to lack of power (n=15 vs n=14).
Up to date, several studies investigating olanzapine-induced changes in glucose metabolism in patients have been published (Hedenmalm et al, 2002; Koller and Doraiswamy, 2002; Sernyak et al, 2002; Wilson et al, 2003; Simpson et al, 2004; Haupt et al, 2007). Newcomer et al (2002) tested oral glucose tolerance in chronically ill BMI-matched patients with schizophrenia. His group found significant glucose elevations (1.0–1.5 SD) in olanzapine-treated patients compared to patients receiving typical antipsychotics as well as to untreated healthy controls.
However, studying metabolic changes in patients can bear difficulties in interpretation. Some authors claim that evidence for insulin resistance, hyperglycemia, and DM2 in patients with major mental disorders are due to an increased prevalence for certain abnormalities in glucose metabolism initially associated with the mental disorder itself and are not necessarily related to any antipsychotic treatment (Mukherjee et al, 1996; Ryan et al, 2003; Cohen et al, 2006). A mechanistic study in dogs proposes a valuable model to examine the effects of atypical antipsychotics in the absence of associated psychiatric disorders (Ader et al, 2005). Ader et al (2005) found that olanzapine-induced weight gain resulted in a dramatic functional impairment in β-cell function compensation compared to fat-fed but unmedicated animals.
As stated in an extensive review article by Bergman and Ader (2005), it is not yet known if results from dogs can be extrapolated to man and agents might differ in their mechanisms of action. To clarify this discussion, we conducted this study in healthy volunteers. Our results are consistent with rodent data (Houseknecht et al, 2005, 2007; Chintoh et al, 2006) as well as similar observations in humans (McIntyre et al, 2001; Simpson et al, 2004). Our current results confirm the previously observed olanzapine-induced changes in glucose metabolism in patients with schizophrenia for healthy volunteers. The time for these metabolic changes to develop in healthy subjects was 10 days of oral intake only, a time period that is shorter than what has been demonstrated for humans before (McIntyre et al, 2001; Newcomer et al, 2002; Simpson et al, 2004).
Nevertheless, it has to be noted that one group has collected controversial data regarding the hypothesis that olanzapine might impair insulin sensitivity in healthy volunteers. Sowell et al (2002) performed hyperglycemic clamps in healthy subjects before and after 3 weeks of oral intake of olanzapine, risperidone or placebo. Despite their finding of substantial weight gain that was reported for olanzapine and risperidone (∼1.5 kg), they did not find the observed changes in whole body insulin sensitivity to be significant. The authors attributed the detected increase in total insulin response for the olanzapine group to the antipsychotic weight gain. But in this study no absolute values of glucose infusion at baseline were reported, which makes it difficult to interpret the results.
In another study, using the euglycemic clamp technique this group failed to detect a significant difference in whole body insulin sensitivity (Sowell et al, 2003). This is surprising but could be partly explained by the partial caloric restriction applied to subjects and by more specific changes in insulin sensitivity of muscle or liver that may have had occurred but were masked because the specific insulin effects were not assessed as hypothesized in Bergman's detailed review on the Sowell studies (Bergman and Ader, 2005).
A clinical study in healthy volunteers bears the drawback of limiting the intake of antipsychotic medication to the minimum period and dosage considered to be absolutely necessary to show first detectable results. This is a limitation of our study, and plasma levels for both ziprasidone and olanzapine were rather low but can still be considered to reach a clinical effective range (Perry et al, 1997; Daniel et al, 1999; Miceli et al, 2000; Mauri et al, 2005). Despite the fact that the doses for olanzapine and ziprasidone are not entirely equivalent in their D(dopamine)2 blocking ability, it is not likely that D2 effects account for the results observed. The doses used are similarly positioned in clinical practice and have been used in many clinical trials (Lieberman et al, 2005; Simpson, 2006).
Neither physical exercise nor food intake restrictions were part of this study design. We did not assess those measures and one can argue that this represents a weakness. However, the subjects were healthy young males who reported not to have changed their exercise or eating patterns during study enrolment.
As shown in Figure 3 the decrease in whole body insulin sensitivity in the olanzapine group clearly becomes more prominent over time. However, it has to be stated that the baseline values of the olanzapine group tend to be higher compared to the ziprasidone baseline values and this difference also develops more prominently over time (olanzapine: 6.5±0.5 vs ziprasidone: 5.4±0.3 ml/h/kg (means±SM) after 100 min of clamp; olanzapine: 6.9±0.5 vs ziprasidone: 5.5±0.3 ml/h/kg (means±SM) after 120 min of clamp). Nevertheless, these differences do not reach statistical significance and cannot sufficiently explain the overall decrease of tolerated glucose uptake in the olanzapine group (baseline: 5.7±0.4 ml/h/kg vs 4.7±0.3 ml/h/kg after 10 days of oral intake, means±SM).
The aim of the study was to investigate potential acute to subacute changes in insulin sensitivity induced by administration of atypical antipsychotics in healthy subjects. In conclusion, our results strongly emphasize the concept that not all atypical antipsychotics cause acute to subacute changes in glucose metabolism and that accurate determination in side-effect profile should be performed when administering an atypical antipsychotic drug. The results do not permit conclusions as to the specific underlying mechanisms behind the development of insulin resistance.
The well-established adiposity-mediated mechanism plays a central role in atypical antipsychotic-induced insulin resistance (American Diabetes Association, 2004b; Casey et al, 2004; Newcomer, 2005; Haupt, 2006; Haupt et al, 2007). Besides unhealthy eating habits of patients with schizophrenia, treatment with atypical antipsychotics seems to increase appetite and worsen opiodergic capacity, thus further deteriorating eating patterns (Elman et al, 2006). There is some evidence that the central nervous system plays an essential role in the development of atypical antipsychotic-induced insulin resistance: whereas one hypothesis proposes modulation of serotonergic/noradrenergic pathways (Kapur and Remington, 2001), another one suggests histamine H1 receptor-linked activation of hypothalamic AMP-kinase (Kim et al, 2007).
On a peripheral level, it has been shown that olanzapine impairs insulin-stimulated glucose transport, increases lipogenesis and inhibits lipolysis in adipocytes (Vestri et al, 2006). Interestingly, a number of studies show evidence for acute, non-adiposity linked mechanisms (Houseknecht et al, 2005) and lack of correlation between weight gain and changes in glucose metabolism (Meyer, 2002; Lindenmayer et al, 2003; Simpson et al, 2004).
Further studies are required to elucidate the specific effects at a cellular level, differentiating between impaired glucose regulation independent of adiposity and weight gain-related effects. Given the still limited knowledge on the cellular pathways involved, Houseknecht et al (2007) argue that experimental models to assess insulin sensitivity are needed to advance our understanding of antipsychotic-induced hyperglycemia, specifically addressing the need for unbiased hyperinsulinemic–euglycemic clamp data in humans. This study aims at addressing that need and our results are consistent with the recently published rodent data by the Houseknecht group. Studies featuring metabolic effects of atypical antipsychotics in normal volunteers are few but essential to provide a chance to assess effects of this medication class on a drug naive population without any potential innate metabolic liabilities of schizophrenia. Even though the current study was partly funded by Pfizer, the study had started before funding was granted, it had been designed and was performed by independent investigators, no company representatives were involved in design, data collection or statistical analysis at any point.
Combining the results on the fast development of first signs of acute to subacute insulin resistance in our study and the facts that are known about progression of DM2 (American Diabetes Association, 2004a), only monitoring fasting glucose levels in patients taking atypical antipsychotics might not be enough. Detailed guidelines before prescribing an atypical antipsychotic have been established (De Hert et al, 2006) and specific recommendations for safe monitoring of weight, blood pressure, blood glucose, and lipid profiles at the beginning and during antipsychotic treatment have been proposed (Newcomer, 2004; Franciosi et al, 2005; Meyer et al, 2005). Switching to an antipsychotic medication with a more appropriate risk/benefit profile has been recommended in patients who gain 5% of their baseline weight at any time during treatment (American Diabetes Association, 2004b).
Even though atypical antipsychotics show essentially equal efficacy, in certain cases a medication switch might not be the best of choices, especially when the originally administered treatment allows optimal control of the patient's symptoms and is otherwise well tolerated (Franciosi et al, 2005). In such a setting, taking additional measures to improve the safety of antipsychotic treatment might be required, such as administering lipid-lowering drugs and antihypertensive medication if lifestyle changes to adopt a healthier diet and increase physical activity are not sufficient (Franciosi et al, 2005).
The results of this study point out the narrow time range that can suffice for antipsychotic-induced acute insulin resistance to develop and clearly show the importance of recognizing any signs of metabolic abnormalities as early as possible to facilitate a successful intervention.
CONFLICT OF INTEREST
Dr Sacher has served as a consultant for Bristol-Myers Squibb, Lundbeck, and Pfizer, Dr Kasper has received grant/research support from Eli Lilly, Lundbeck, Bristol-Myers Squibb, GlaxoSmithKline, Organon, Pfizer, Sepracor, and Servier; has served as a consultant or on advisory boards for AstraZeneca, Bristol-Myers Squibb, Glaxo-SmithKline, Eli Lilly, Lundbeck, Pfizer, Organon, Sepracor, Janssen, and Novartis; and has served on speakers' bureaus for AstraZeneca, Eli Lilly, Lundbeck, Sepracor, and Janssen.
References
Ader M, Kim SP, Catalano KJ, Ionut V, Hucking K, Richey JM et al (2005). Metabolic dysregulation with atypical antipsychotics occurs in the absence of underlying disease: a placebo-controlled study of olanzapine and risperidone in dogs. Diabetes 54: 862–871.
American Diabetes Association (2004a). Diagnosis and classification of diabetes mellitus. Diabetes Care 27 (Suppl 1): S5–S10.
American Diabetes Association, A.P.A., American Association of Clinical Endocrinologists, North American Association for the Study of Obesity (2004b). Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 27: 596–601.
Bergman RN (1989). Lilly lecture 1989. Toward physiological understanding of glucose tolerance. Minimal-model approach. In Diabetes. Vol. 3, Text No. 12, pp 1512–1527 (http://diabetes.diabetesjournals.org/cgi/content/abstract/38/12/1512).
Bergman RN, Ader M (2005). Atypical antipsychotics and glucose homeostasis. J Clin Psychiatry 66: 504–514.
Bleuler M (1951). Some aspects of endocrinologic psychiatry. J Nerv Ment Dis 113: 74–76.
Boehm G, Racoosin JA, Laughren TP, Katz R (2004). Consensus development conference on antipsychotic drugs and obesity and diabetes: response to consensus statement. Diabetes Care 27: 2088–2089; author reply 2089–2090.
Casey DE, Haupt DW, Newcomer JW, Henderson DC, Sernyak MJ, Davidson M et al (2004). Antipsychotic-induced weight gain and metabolic abnormalities: implications for increased mortality in patients with schizophrenia. J Clin Psychiatry 65 (Suppl 7): 4–18; quiz 19–20.
Chintoh AMS, Lam S, Lam T, Naasasan A, Remington G, Giacca A (2006). Insulin Resistance following olanzapine administration: an animal Model. Int J Neuropsychopharmacol 9: S144.
Cohen D, Stolk RP, Grobbee DE, Gispen-de Wied CC (2006). Hyperglycemia and diabetes in patients with schizophrenia or schizoaffective disorders. Diabetes Care 29: 786–791.
Conley RR, Tamminga CA, Bartko JJ, Richardson C, Peszke M, Lingle J et al (1998). Olanzapine compared with chlorpromazine in treatment-resistant schizophrenia. Am J Psychiatry 155: 914–920.
Daniel DG, Zimbroff DL, Potkin SG, Reeves KR, Harrigan EP, Lakshminarayanan M (1999). Ziprasidone 80 mg/day and 160 mg/day in the acute exacerbation of schizophrenia and schizoaffective disorder: a 6-week placebo-controlled trial. Ziprasidone Study Group. Neuropsychopharmacol 20: 491–505.
De Hert M, van Eyck D, De Nayer A (2006). Metabolic abnormalities associated with second generation antipsychotics: fact or fiction? Development of guidelines for screening and monitoring. Int Clin Psychopharmacol 21 (Suppl 2): S11–S15.
DeFronzo RA, Tobin JD, Andres R (1979). Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237: E214–E223.
Dixon L, Weiden P, Delahanty J, Goldberg R, Postrado L, Lucksted A et al (2000). Prevalence and correlates of diabetes in national schizophrenia samples. Schizophr Bull 26: 903–912.
Elman I, Borsook D, Lukas SE (2006). Food intake and reward mechanisms in patients with schizophrenia: implications for metabolic disturbances and treatment with second-generation antipsychotic agents. Neuropsychopharmacology 31: 2091–2120.
FDA (2004). MedWatch Safety Info. In Previous News Items (see http://www.fda.gov/cder/previous_news2004.htm, MedWatch Report, 13 April 2004).
Franciosi L, Kasper S, Garber A, Johnson D, Krauss R, Marder S et al (2005). Advancing the treatment of people with mental illness: A call to action in the management of metabolic issues. J Clin Psychiatry 66: 790–798.
Gianfrancesco F, Grogg A, Mahmoud R, Wang RH, Meletiche D (2003). Differential effects of antipsychotic agents on the risk of development of type 2 diabetes mellitus in patients with mood disorders. Clin Ther 25: 1150–1171.
Haupt DW (2006). Differential metabolic effects of antipsychotic treatments. Eur Neuropsychopharmacol 16 (Suppl 3): S149–S155.
Haupt DW, Newcomer JW (2002). Abnormalities in glucose regulation associated with mental illness and treatment. J Psychosom Res 53: 925–933.
Haupt DW, Fahnestock PA, Flavin KA, Schweiger JA, Stevens A, Hessler MJ et al (2007). Adiposity and insulin sensitivity derived from intravenous glucose tolerance tests in antipsychotic-treated patients. Neuropsychopharmacology (advance online publication, 21 March 2007; doi:10.1038/sj.npp.1301392).
Hedenmalm K, Hagg S, Stahl M, Mortimer O, Spigset O (2002). Glucose intolerance with atypical antipsychotics. Drug Saf 25: 1107–1116.
Henderson DC, Cagliero E, Copeland PM, Borba CP, Evins E, Hayden D et al (2005). Glucose metabolism in patients with schizophrenia treated with atypical antipsychotic agents: a frequently sampled intravenous glucose tolerance test and minimal model analysis. Arch Gen Psychiatry 62: 19–28.
Henderson DC, Copeland PM, Borba CP, Daley TB, Nguyen DD, Cagliero E et al (2006). Glucose metabolism in patients with schizophrenia treated with olanzapine or quetiapine: a frequently sampled intravenous glucose tolerance test and minimal model analysis. J Clin Psychiatry 67: 789–797.
Holmang A, Muller M, Andersson OK, Lonnroth P (1998). Minimal influence of blood flow on interstitial glucose and lactate-normal and insulin-resistant muscle. Am J Physiol 274: E446–E452.
Houseknecht KL, Robertson AS, Zavadoski W, Gibbs EM, Johnson DE, Rollema H (2007). Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: implications for adverse metabolic effects. Neuropsychopharmacology 32: 289–297.
Houseknecht KL, Robertson AS, Johnson DE, Rollema H (2005). Clozapine and olanzapine, but not ziprasidone, cause acute insulin resistance in normal rats. Biol Psychiatry 57: 128.
Jansson PA, Larsson A, Smith U, Lonnroth P (1992). Glycerol production in subcutaneous adipose tissue in lean and obese humans. J Clin Invest 89: 1610–1617.
Kane JM, Barrett EJ, Casey DE, Correll CU, Gelenberg AJ, Klein S et al (2004). Metabolic effects of treatment with atypical antipsychotics. J Clin Psychiatry 65: 1447–1455.
Kapur S, Remington G (2001). Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry 50: 873–883.
Karter AJ, Mayer-Davis EJ, Selby JV, D'Agostino Jr RB, Haffner SM, Sholinsky P et al (1996). Insulin sensitivity and abdominal obesity in African-American, Hispanic, and non-Hispanic white men and women. The Insulin Resistance and Atherosclerosis Study. Diabetes 45: 1547–1555.
Kasper S, Hale A, Azorin JM, Moller HJ (1999). Benefit-risk evaluation of olanzapine, risperidone and sertindole in the treatment of schizophrenia. Eur Arch Psychiatry Clin Neurosci 249 (Suppl 2): II1–II14.
Kasper S, Rosillon D, Duchesne I (2001). Risperidone olanzapine drug outcomes studies in schizophrenia (RODOS): efficacy and tolerability results of an international naturalistic study. Int Clin Psychopharmacol 16: 179–187.
Kim FS, Huang A, Snowman MA, Teuscher C, Snyder HS (2007). Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc Natl Acad Sci USA 104: 3456–3459.
Kinon BJ, Basson BR, Gilmore JA, Tollefson GD (2001). Long-term olanzapine treatment: weight change and weight-related health factors in schizophrenia. J Clin Psychiatry 62: 92–100.
Koller EA, Doraiswamy PM (2002). Olanzapine-associated diabetes mellitus. Pharmacotherapy 22: 841–852.
Kopelowicz A, Zarate R, Tripodis K, Gonzalez V, Mintz J (2000). Differential efficacy of olanzapine for deficit and nondeficit negative symptoms in schizophrenia. Am J Psychiatry 157: 987–993.
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO et al (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353: 1209–1223.
Lillioja S, Bogardus C (1988). Obesity and insulin resistance: lessons learned from the Pima Indians. Diabetes Metab Rev 4: 517–540.
Lindenmayer JP, Czobor P, Volavka J, Citrome L, Sheitman B, McEvoy JP et al (2003). Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry 160: 290–296.
Macor C, Ruggeri A, Mazzonetto P, Federspil G, Cobelli C, Vettor R (1997). Visceral adipose tissue impairs insulin secretion and insulin sensitivity but not energy expenditure in obesity. Metabolism 46: 123–129.
Mauri MC, Steinhilber CP, Marino R, Invernizzi E, Fiorentini A, Cerveri G et al (2005). Clinical outcome and olanzapine plasma levels in acute schizophrenia. Eur Psychiatry 20: 55–60.
Mayer-Davis EJ, Monaco JH, Hoen HM, Carmichael S, Vitolins MZ, Rewers MJ et al (1997). Dietary fat and insulin sensitivity in a triethnic population: the role of obesity. The Insulin Resistance Atherosclerosis Study (IRAS). Am J Clin Nutr 65: 79–87.
McIntyre RS, Mancini DA, Basile VS (2001). Mechanisms of antipsychotic-induced weight gain. J Clin Psychiatry 62 (Suppl 23): 23–29.
Meyer J, Koro CE, L'Italien GJ (2005). The metabolic syndrome and schizophrenia: a review. Int Rev Psychiatry 17: 173–180.
Meyer JM (2002). A retrospective comparison of weight, lipid, and glucose changes between risperidone- and olanzapine-treated inpatients: metabolic outcomes after 1 year. J Clin Psychiatry 63: 425–433.
Miceli JJ, Wilner KD, Hansen RA, Johnson AC, Apseloff G, Gerber N (2000). Single- and multiple-dose pharmacokinetics of ziprasidone under non-fasting conditions in healthy male volunteers. Br J Clin Pharmacol 49 (Suppl 1): 5S–13S.
Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP (2001). The continuing epidemics of obesity and diabetes in the United States. Jama 286: 1195–1200.
Mukherjee S, Decina P, Bocola V, Saraceni F, Scapicchio PL (1996). Diabetes mellitus in schizophrenic patients. Compr Psychiatry 37: 68–73.
Muller M (2002). Science, medicine, and the future: microdialysis. BMJ 324: 588–591.
Muller M, Schmid R, Georgopoulos A, Buxbaum A, Wasicek C, Eichler HG (1995). Application of microdialysis to clinical pharmacokinetics in humans. Clin Pharmacol Ther 57: 371–380.
Newcomer JW (2004). Metabolic risk during antipsychotic treatment. Clin Ther 26: 1936–1946.
Newcomer JW (2005). Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 19 (Suppl 1): 1–93.
Newcomer JW, Haupt DW, Fucetola R, Melson AK, Schweiger JA, Cooper BP et al (2002). Abnormalities in glucose regulation during antipsychotic treatment of schizophrenia. Arch Gen Psychiatry 59: 337–345.
Okamura F, Tashiro A, Utumi A, Imai T, Suchi T, Tamura D et al (2000). Insulin resistance in patients with depression and its changes during the clinical course of depression: minimal model analysis. Metabolism 49: 1255–1260.
Perry PJ, Sanger T, Beasley C (1997). Olanzapine plasma concentrations and clinical response in acutely ill schizophrenic patients. J Clin Psychopharmacol 17: 472–477.
Ryan MC, Collins P, Thakore JH (2003). Impaired fasting glucose tolerance in first-episode, drug-naive patients with schizophrenia. Am J Psychiatry 160: 284–289.
Sanger TM, Lieberman JA, Tohen M, Grundy S, Beasley Jr C, Tollefson GD (1999). Olanzapine versus haloperidol treatment in first-episode psychosis. Am J Psychiatry 156: 79–87.
Sernyak MJ, Leslie DL, Alarcon RD, Losonczy MF, Rosenheck R (2002). Association of diabetes mellitus with use of atypical neuroleptics in the treatment of schizophrenia. Am J Psychiatry 159: 561–566.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al (1998). The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59 (Suppl 20): 22–33; quiz 34–57.
Simpson GM (2006). Efficacy and tolerability of ziprasidone and olanzapine in acutely ill inpatients with schizophrenia or schizoaffective disorder: results of a double-blind, six-week study, with a six-month, double-blind continuation Phase. Prog Neurotherapeutics Neuropsychopharmacol 1: 149–163.
Simpson GM, Glick ID, Weiden PJ, Romano SJ, Siu CO (2004). Randomized, controlled, double-blind multicenter comparison of the efficacy and tolerability of ziprasidone and olanzapine in acutely ill inpatients with schizophrenia or schizoaffective disorder. Am J Psychiatry 161: 1837–1847.
Sowell M, Mukhopadhyay N, Cavazzoni P, Carlson C, Mudaliar S, Chinnapongse S et al (2003). Evaluation of insulin sensitivity in healthy volunteers treated with olanzapine, risperidone, or placebo: a prospective, randomized study using the two-step hyperinsulinemic, euglycemic clamp. J Clin Endocrinol Metab 88: 5875–5880.
Sowell MO, Mukhopadhyay N, Cavazzoni P, Shankar S, Steinberg HO, Breier A et al (2002). Hyperglycemic clamp assessment of insulin secretory responses in normal subjects treated with olanzapine, risperidone, or placebo. J Clin Endocrinol Metab 87: 2918–2923.
Unwin N, Shaw J, Zimmet P, Alberti KG (2002). Impaired glucose tolerance and impaired fasting glycaemia: the current status on definition and intervention. Diabet Med 19: 708–723.
Vestri HS, Maianu L, Moellering DR, Garvey WT (2006). Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology 32: 765–772.
Welch S, Gebhart SS, Bergman RN, Phillips LS (1990). Minimal model analysis of intravenous glucose tolerance test-derived insulin sensitivity in diabetic subjects. J Clin Endocrinol Metab 71: 1508–1518.
Wilson DR, D'Souza L, Sarkar N, Newton M, Hammond C (2003). New-onset diabetes and ketoacidosis with atypical antipsychotics. Schizophr Res 59: 1–6.
Acknowledgements
The study was funded by ‘Jubilaeumsfond Oesterreichische National bank’ (no. 11067) and a research grant from Pfizer. We express our sincere gratitude to Julia Szendroedi, MD and Rupert Lanzenberger, MD, for helpful discussion and critical advice, as well as to cand med Lukas Zinnagl and Petra Zeleny for their assistance in performing the hyperinsulinemic euglycemic clamps.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sacher, J., Mossaheb, N., Spindelegger, C. et al. Effects of Olanzapine and Ziprasidone on Glucose Tolerance in Healthy Volunteers. Neuropsychopharmacol 33, 1633–1641 (2008). https://doi.org/10.1038/sj.npp.1301541
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301541
Keywords
This article is cited by
-
Antipsychotic-Induced Weight Gain in Severe Mental Illness: Risk Factors and Special Considerations
Current Psychiatry Reports (2023)
-
Insulin and glucose metabolism with olanzapine and a combination of olanzapine and samidorphan: exploratory phase 1 results in healthy volunteers
Neuropsychopharmacology (2022)
-
Acute Hyperglycemia Associated with Short-Term Use of Atypical Antipsychotic Medications
Drugs (2014)
-
Management of Medication-Related Cardiometabolic Risk in Patients with Severe Mental Illness
Current Cardiovascular Risk Reports (2013)
-
Atypical antipsychotics and diabetic ketoacidosis: a review
Psychopharmacology (2013)
