
Abstract
Study design:
Lithium has attracted much attention as a neuroregenerative agent for spinal cord injury in animal models. We hypothesized that the lithium can be beneficial to patients with spinal cord injury. The safety and pharmacokinetics of lithium has been studied in our earlier phase I clinical trial, indicating its safety. This is a phase II clinical trial to evaluate its efficacy on chronic spinal cord injury patients.
Objectives:
The aim of this study was to investigate the efficacy of lithium on chronic spinal cord injury patients.
Setting:
A major spinal cord injury rehabilitation center in Beijing, China.
Methods:
Randomized, double-blind, placebo-controlled 6-week parallel treatment arms with lithium carbonate and with placebo. A total of 40 chronic spinal cord injury subjects were recruited. Oral lithium carbonate was titrated or placebo was simulated to maintain the serum lithium level of 0.6–1.2 mmol l−1 for 6 weeks, followed by a 6-month follow-up. The functional outcomes and the neurological classifications, as well as the safety parameters, adverse events and pharmacokinetic data were carefully collected and monitored.
Results:
No significant changes in the functional outcomes and the neurological classifications were found. The only significant differences were in the pain assessments using visual analog scale comparing the lithium and the placebo group. No severe adverse event was documented in the study.
Conclusion:
The lithium treatment did not change the neurological outcomes of patients with chronic spinal cord injury. It is worth to investigate whether lithium is effective in the treatment of neuropathic pain in chronic spinal cord injury.
Sponsorship:
China Spinal Cord Injury Network Company Limited.
Similar content being viewed by others
Introduction
Lithium attracted much attention as a potential neuroregenerative agent for spinal cord injury in 2004 when Yick et al.1 reported that lithium treatment reinforced regeneration-promoting effects of the chondroitinase ABC on rat rubrospinal neurons. In 2011, Wong et al.2 reported the safety and pharmacokinetic study of lithium in chronic spinal cord injury patients. Wong's investigation is the first step of series of clinical trials supported by the China Spinal Cord Injury Network to assess the effects of umbilical cord blood and lithium on chronic spinal cord injury. We designed the current phase II clinical trial to investigate the safety and efficacy of the 6-week oral lithium carbonate treatment of chronic spinal cord injury.
Materials and methods
This study is a prospective, randomized, double-blind, placebo-controlled efficacy trial to evaluate the effects of lithium carbonate on neurological findings and functional outcomes in patients with stable chronic spinal cord injury. We defined stable chronic spinal cord injury as 12 months or longer after injury with no change in American Spinal Injury Association (ASIA) Classification of A, B or C for at least 6 months. The Institutional Review Board of China Rehabilitation Research Centre (CRRC) approved the study protocol, and informed consent and the trial was registered at www.clinicaltrials.gov (NCT00750061). A total of 40 eligible patients were recruited from CRRC, Beijing from 2008 to 2009. The investigation adhered to the principles of the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice. All patients gave written informed consent before their enrollment.
Subject population
Patients, 18–60 years old, with C4 to T10 spinal cord injury more than 12 months and ASIA impairment scale of A to C unchanged for more than 6 months were eligible. Exclusion criteria included: hypersensitivity to lithium; significant renal, cardiovascular, hepatic, psychiatric or other medical diseases; associated severe brain injury; concomitant intake of drugs interacting with lithium; history of alcohol or drug abuse; and pregnant or lactating women and women reluctant for effective contraception.
Study design and drug administration
A total of 40 eligible patients were randomized into two groups. The subjects in the Treatment Group received lithium carbonate, while the Control Group received placebo. Neither the subjects nor the physicians knew which group the subjects were allocated to. Each subject received oral lithium carbonate or placebo for 6 weeks. The dose was adjusted according to the serum lithium level report, the real lithium report in the Treatment Group and a sham report in the Control Group. The outcomes were assessed 6 weeks and 6 months after the onset of the medication, and were compared with baseline pre-treatment data to obtain ‘neurological change scores’. The efficacy and safety were analyzed comparing the results of the Treatment Group with those of the Control Group.
Dose modification
The dosing regimen aimed at achieving therapeutic level of lithium is 0.6–1.2 mmol l−1. Dose titration followed the schedule shown in Figure 1. All the enrolled subjects began with three tablets (250 mg lithium carbonate per tablet or placebo) daily. Seventy-two hours (day 3) after the initial oral intake, blood was sampled and serum was shipped to a designated center to measure lithium concentration. The actual serum lithium level was reported if the subject was in Lithium Group whereas a pre-scheduled sham result, based on our previous study, was reported for subjects on placebo. The reporter of the lithium test was independent and non-blinded regarding the treatment grouping. The investigator was advised to adjust the daily dosage based on the report according to the following scheme if the clinical condition allowed:

Dosage Titration Suggestion.
If the serum lithium concentration reached the level of 0.6–1.2 mmol l−1, no adjustment of dosage was required. The same dosage was maintained for 6 weeks. If the initial serum lithium at day 3 did not reach the minimum level of 0.6 mmol l−1, an additional 250 mg tablet was added. Blood was sampled at week 1. If the minimum level 0.6 mmol l−1 was still not achieved, an additional 250 mg tablet would be added and blood was sampled again at 72 h post-dosage adjustment. This was repeated until the criterion of 0.6–1.2 mmol l−1 was reached.
If the lithium levels exceeded the maximum level of 1.2 mmol l−1 and below 1.5 mmol l−1 without presenting any symptoms of intoxication at day 3, the dosage was reduced by one tablet, that is, 250 mg. Blood for lithium was taken at week 1. If the serum concentration still exceeded the maximum level, the subject was withdrawn from the study and serum lithium level was determined 5 days afterward. If the minimum level of 0.6 mmol l−1 was reached, no further readjustment of drug dosage was required and the subject was maintained at that dosage level for the remainder of the 6-week treatment period.
Efficacy evaluation
The primary study endpoints were the changes in the neurological scoring, from baseline (Visit 2) to week 6 (Visit 4) and month 6 (Visit 5), specifically: (a) Means of changed motor scores, (b) Means of changed sensory scores (light touch and pinprick) and (c) ASIA Impairment Scale grading change.
The secondary endpoints were the changes in the following parameters from baseline to week 6 and month 6: (a) Functional Independence Measure (FIM), (b) Visual Analog Scale (VAS)—Pain and (c) Modified Ashworth Scale (MAS)—Spasticity.
Safety evaluations
Safety profile included adverse events, vital signs, electrocardiogram, routine blood and urine tests, hepatic, renal and thyroid function tests, and neurological assessments.
All patients were monitored for adverse events. Clinical assessment included side effects of lithium, body weight, vital signs (temperature, pulses, respiratory rates, blood pressures in the sitting position), VAS for any pain all over the body, Ashworth scale of limbs below the neurological level and ASIA scoring. Scheduled blood tests were done to monitor the renal, liver, thyroid and parathyroid functions. In addition, full blood count, clotting profile, serum glucose level and urinalysis were performed to document any other possible adverse events. Electrocardiography was used to monitor cardiac status.
Statistical analyses
Sample size calculation was estimated on the basis of motor score changes, using the method of Zhao.3 On the basis of our observational study, the required total sample size to achieve an 80% power (β=0.2) at 5% probability of type I error (α=0.05) for correctly detecting such difference of 5 motor score change is 18. On the basis of this calculation, this trial enrolled 40 subjects in total, 20 subjects per group.
Primary and secondary efficacy analyses were performed on the per-protocol set, the dataset that has excluded the cases of poor compliance, dropouts and so on. The safety analysis set consists of the subjects who have taken at least one tablet of the investigational product.
All statistical tests were two-tailed, deemed significant when P<0.05. The light touch score, pin prick score, motor score and VAS (for pain) were tested with Wilcoxon rank sum; the ASIA Impairment Scale, MAS (for spasticity) were tested with Cochran–Mantel–Haenszel statistics and the FIM motor score was tested with Student's t-test. The safety analysis was comparing the incidence of subjects with adverse events and the incidence of adverse events between the two groups.
Data locking and unblinding
After data input, verification and confirmation, the first-time unblinding was done, allowing the statistician to know the group assignment of each subject and to compare the data of the two groups statistically.
After the statistical summary is confirmed, the second unblinding was done, allowing the summary reporter to know whether Group A or Group B is Lithium Group or Placebo Group in order to write the trial report. In this trial, Groups A and B were, respectively, Placebo and Lithium Groups.
Results
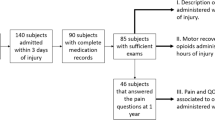
A total of 40 eligible subjects were randomly allocated to receive lithium carbonate (250 mg per tablet) or placebo for 6 weeks. Among the 40 subjects, 20 were treated with lithium carbonate and 20 with placebo.
Table 1 and 2 compare demographic data and etiology. The randomization achieved balance of observed demographic characteristics and injury etiology between the two treatment groups. Two (10%) patients in each group dropped out of the study. Table 3 lists the reasons for the dropout as being due to adverse events.
Primary outcomes
Figures 2, 3 and 4 show the motor scores, sensory scores (light touch and pin prick) and ASIA impairment scales (AIS), respectively. Baseline motor score, pin prick score and AIS did not differ between treatment groups (36.28±20.375 vs 45.22±14.591, 38.39±21.704 vs 51.67±21.601 and 15A/1B/2C vs 14A/2B/2C in lithium carbonate and placebo groups, respectively; P=0.119, 0.051 and 0.804; per-protocol set).

Motor Scores.

Sensory Scores.

Change of ASIA Impairment Scale at visit 2, visit 4 and visit 5.
The baseline light touch scores are a little different. At the beginning, the baseline scores did not differ between the two groups (53.80±20.786 vs 43.45±23.091 in lithium carbonate and placebo groups, respectively, P=0.081, full analysis set), but after the four subjects dropped out, the baseline scores became significantly different (54.44±20.267 vs 40.72±22.741 in lithium carbonate and placebo groups, respectively, P=0.040, per-protocol set).
The changes of motor scores, light touch scores, pin prick scores and AIS from after 6 weeks intervention therapy and 6 months follow-up were small in both lithium carbonate and placebo groups. The difference of the changes between the two groups is not of statistical significance.
Secondary outcomes
The FIM motor scores, the VASs for pain and the MASs did not differ between the randomized groups at baseline.
FIM motor score and the MAS for spasticity did not change after 6 weeks intervention therapy and at the 6 months follow-up in both groups. The difference between the two groups was not significant.
After 6 weeks of treatment, VAS pain scores in the lithium carbonate group were significantly greater than those in placebo group (−1±3.97 vs 8.833±14.861; P=0.034; Figure 5). This effect lasted for 6 months (Figure 6), 4 and half months after discontinuation of lithium therapy (0.778±17.176 vs 9.389±15.232; P=0.041).

ΔVAS pain score stands for the change of the VAS for pain scores. FAS, full analysis set; PPS, per-protocol set.

Four patients in Group B (B10, B17, B18 and B19) had obvious reduction of VASs at 6 weeks and 6 months. In Group A, only one subject had reduced neuropathic pain, but this was after the 6-week treatment period.
Safety and tolerability
The average serum lithium level of the lithium group after 3 days was 0.54 mmol l−1±0.152. (n=19) and was 0.68 mmol l−1±0.155 (n=18) after 6 weeks. During the 6 weeks of intervention and 6 months follow-up, 16 subjects (80%) in the lithium carbonate and 14 subjects (70%) in the placebo group reported at least one adverse event (P=0.465). The total number of adverse event reports differed between study groups: 56 versus 30 events in the lithium carbonate and placebo groups, respectively (P=0.007). The common adverse event in the lithium group was nausea. No server side effects causing organ dysfunction were observed within the therapeutic lithium level.
Discussion
Lithium stimulates proliferation of many types of stem cells, including neural stem cells,4, 5 mesenchymal stem cells,6, 7 hematopoietic stem cells8, 9, 10 and embryonic stem cells.11, 12 Lithium also stimulates T cells13 and enhances survival of neural stem cells.14 Thus, lithium has many effects that are potentially neuroregenerative for spinal cord injury. In 2011, Wong et al.2 reported that lithium can be safely given to chronic spinal cord injury patients and can be maintained in a therapeutic range of 0.6–1.2 mmol l−1 over a 6-week period. We consequently designed the current phase II clinical trial to assess the effectiveness of 6-week oral lithium carbonate treatment of chronic spinal cord injury.
In this double-blind, randomized trial, lithium did not change motor scores, sensory scores, ASIA Impairment Scales, FIM motor scores or the MAS for spasticity in 20 patients with chronic spinal cord injury compared with 20 patients who received placebo. This is not surprising, considering that regenerative therapies must ‘bridge’ the injury site, provide sustained growth factor support and inhibit axonal growth inhibitors known to be present in the spinal cord around the injury site. An individual therapy such as oral lithium may not be sufficient to regenerate the spinal cord.
We worried that lithium may increase neuropathic pain in patients with chronic spinal cord injury. Neurotrophins have been implicated in neuropathic pain, particularly brain-derived neurotrophic factor15 and nerve growth factor.16 In our study, about half of the subjects had severe neuropathic pain (VAS scores >50/100). To our surprise, the 6-week course of oral lithium carbonate treatment markedly reduced VAS scores, not only at the end of the 6-week treatment period but also at the 6 months follow-up examination, 4 and a half months after the lithium was stopped. Lithium eliminated severe neuropathic pain of two patients, one with a thoracic and the other with cervical spinal cord injury.
If this effect of lithium can be confirmed by further investigation, what are some potential mechanisms? In 2000, Shimizu et al.17 reported that intrathecal lithium reduces neuropathic pain responses in a rat model of peripheral neuropathy. Lithium is used clinically to treat cluster headaches.18, 19, 20, 21 However, lithium is not considered an analgesic,22 does not activate opioid receptors23 and may even aggravate pain by antagonizing cholecystokinin octapeptide reversal of opioid effects24 that may contribute to neuropathic pain after spinal cord injury.25 Lithium even facilitates endotoxin-mediated hyperalgesia behavior.26 Thus, lithium is unlikely to be acting through analgesic or a pain-behavior suppression mechanism.
Several recent studies suggest that increasing neurotrophin levels in spinal cord injury or other neuropathic pain models may even alleviate neuropathic pain. For example, NGF,27 BDNF28 and NT329, 30 all reduce neuropathic pain under certain conditions. It is also possible that neurotrophins need to be associated with inflammation in order to cause neuropathic pain. For example, Chen et al.31 found that immune activation is required for NT-3-induced axonal plasticity in chronic spinal cord injury. Finally, some neurotrophins may suppress sprouting. Ramer et al.32 found that endogenous TrkB ligands suppress sprouting and mechanosensory recovery in rats after dorsal root injuries.
Lithium acts by inhibiting glycogen synthetase kinase 3-beta (GSK3B),33 which in turn inhibits nuclear factors that upregulate growth and survival genes in cells,34 including the nuclear factor of activated T cells (NFAT) and Wnt.35 Is GSK3β inhibition associated with relief of neuropathic pain?
Xie et al.36 demonstrated that morphine-induced apoptosis in microglial cells is mediated via GSK-3beta and p38 MAPK pathways, while targeting microglial signaling might lead to more effective treatments for devastating chronic pain.37 Parkitna et al.38 reported that a single intrathecal injection of GSK3β inhibitor can restore the analgesic effect of morphine in morphine-tolerant rats.
Should the antidepressive effect of lithium explain its relief of chronic pain? Pain and depression are closely related. Depression can cause pain and pain can cause depression. Sometimes pain and depression create a vicious cycle in which pain worsens symptoms of depression, and then the resulting depression worsens feelings of pain.39 However, this reason cannot explain the effect lasting 1 and a half months after stopping medication.
In retrospect, we perhaps should not have worried so much that lithium may increase neuropathic pain. Lithium is still the most effective therapy for manic depression.40 Millions of people take lithium, often for a lifetime.41 As many people who take lithium also have neuropathic pain, if lithium aggravated or caused neuropathic pain, such effects would have been reported by now. At serum levels of 0.6–1.2 mmol l−1,42 the side effects of lithium are well known,40 including gastrointestinal pain or discomfort, diarrhea, tremor, polyuria, nocturnal urination, weight gain, edema, flattening of affect and dermatological eruptions.43 In our study, lithium did not increase visual analog scores in any patient with or without pre-existing neuropathic pain.
In summary, our study confirmed that people with chronic spinal cord injury tolerated a 6-week course of oral lithium carbonate treatment well and they showed no evidence of organ dysfunction associated with 0.6–1.2 mmol l−1 blood levels of lithium for 6 weeks. The lithium treatment did not change the neurological outcomes, either in comparison with the control group or relative to the patients’ pre-injury scores. However, the treatment reduced neuropathic pain during the 6-week treatment period and appeared to have continuing effects for several months even after the lithium was discontinued.
Data Archiving
There was no data to deposit.
References
Yick LW, So KF, Cheung PT, Wu WT . Lithium chloride reinforces the regeneration-promoting effect of chondroitinase ABC on rubrospinal neurons after spinal cord injury. J Neurotrauma 2004; 21: 932–943.
Wong YW, Tam S, So KF, Chen JY, Cheng WS, Luk KD et al. A three-month, open-label, single-arm trial evaluating the safety and pharmacokinetics of oral lithium in patients with chronic spinal cord injury. Spinal Cord 2011; 49: 94–98.
Zhao YD, Rahardja D, Qu Y . Sample size calculation for the Wilcoxon-Mann-Whitney test adjusting for ties. Stat Med 2008; 27: 462–468.
Wexler EM, Geschwind DH, Palmer TD . Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry 2008; 13: 285–292.
Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang DM . Lithium stimulates progenitor proliferation in cultured brain neurons. Neuroscience 2003; 117: 55–61.
De Boer J, Wang HJ, Van Blitterswijk C . Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng 2004; 10: 393–401.
De Boer J, Siddappa R, Gaspar C, van Apeldoorn A, Fodde R, van Blitterswijk C . Wnt signaling inhibits osteogenic differentiation of human mesenchymal stem cells. Bone 2004; 34: 818–826.
Levitt LJ, Quesenberry PJ . The effect of lithium on murine hematopoiesis in a liquid culture system. N Engl J Med 1980; 302: 713–719.
Aglietta M, Leone A, Piacibello W . Effect of lithium on normal and chronic granulocytic leukemia colony forming cells (CFU-GM). Experientia 1981; 37: 1340–1341.
Gallicchio VS, Chen MG . Influence of lithium on proliferation of hematopoietic stem cells. Exp Hematol 1981; 9: 804–810.
Anton R, Kestler HA, Kuhl M . Beta-catenin signaling contributes to stemness and regulates early differentiation in murine embryonic stem cells. FEBS Lett 2007; 581: 5247–5254.
Schmidt MM, Guan K, Wobus AM . Lithium influences differentiation and tissue-specific gene expression of mouse embryonic stem (ES) cells in vitro. Int J Dev Biol 2001; 45: 421–429.
Ohteki T, Parsons M, Zakarian A, Jones RG, Nguyen LT, Woodgett JR et al. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J Exp Med 2000; 192: 99–104.
Willing AE, Zigova T, Milliken M, Poulos S, Saporta S, McGrogan M et al. Lithium exposure enhances survival of NT2N cells (hNT neurons) in the hemiparkinsonian rat. Eur J Neurosci 2002; 16: 2271–2278.
Vanelderen P, Rouwette T, Kozicz T, Roubos E, Van Zundert J, Heylen R et al. The role of brain-derived neurotrophic factor in different animal models of neuropathic pain. Eur J Pain 2010; 14: 473 e1-9.
Jing YY, Wang JY, Li XL, Wang ZH, Pei L, Pan MM et al. Nerve growth factor of red nucleus involvement in pain induced by spared nerve injury of the rat sciatic nerve. Neurochem Res 2009; 34: 1612–1618.
Shimizu T, Shibata M, Wakisaka S, Inoue T, Mashimo T, Yoshiya I . Intrathecal lithium reduces neuropathic pain responses in a rat model of peripheral neuropathy. Pain 2000; 85: 59–64.
Jurgens TP, Schaefer C, May A . Treatment of cluster headache in pregnancy and lactation. Cephalalgia 2009; 29: 391–400.
Matharu M, Silver N . Cluster headache. Clin Evid (Online) 2008.
Leroux E, Ducros A . Cluster headache. Orphanet J Rare Dis 2008; 3: 20.
Loder E . Prophylaxis: headaches that never happen. Headache 2008; 48: 694–696.
Karakucuk EH, Yamanoglu T, Demirel O, Bora N, Zengil H . Temporal variation in drug interaction between lithium and morphine-induced analgesia. Chronobiol Int 2006; 23: 675–682.
Johnston IN, Westbrook RF . Inhibition of morphine analgesia by lithium: role of peripheral and central opioid receptors. Behav Brain Res 2004; 151: 151–158.
Zhang LJ, Han NL, Han JS . Regulation by lithium of the antagonistic effect of cholecystokinin octapeptide on ohmefentanyl-induced antinociception. Neuropharmacology 1994; 33: 123–126.
Kim J, Kim JH, Kim Y, Cho HY, Hong SK, Yoon YW . Role of spinal cholecystokinin in neuropathic pain after spinal cord hemisection in rats. Neurosci Lett 2009; 462: 303–307.
Maier SF, Wiertelak EP, Martin D, Watkins LR . Interleukin-1 mediates the behavioral hyperalgesia produced by lithium chloride and endotoxin. Brain Res 1993; 623: 321–324.
Cirillo G, Cavaliere C, Bianco MR, De Simone A, Colangelo AM, Sellitti S et al. Intrathecal NGF administration reduces reactive astrocytosis and changes neurotrophin receptors expression pattern in a rat model of neuropathic pain. Cell Mol Neurobiol 2010; 30: 51–62.
Eaton MJ, Blits B, Ruitenberg MJ, Verhaagen J, Oudega M . Amelioration of chronic neuropathic pain after partial nerve injury by adeno-associated viral (AAV) vector-mediated over-expression of BDNF in the rat spinal cord. Gene Ther 2002; 9: 1387–1395.
Tender GC, Kaye AD, Li YY, Cui JG . Neurotrophin-3 and Tyrosine-Kinase C Have Modulatory Effects on Neuropathic Pain in the Rat Dorsal Root Ganglia. Neurosurgery 2011; 68: 1048–1055.
Wilson-Gerwing TD, Stucky CL, McComb GW, Verge VM . Neurotrophin-3 significantly reduces sodium channel expression linked to neuropathic pain states. Exp Neurol 2008; 213: 303–314.
Chen Q, Smith GM, Shine HD . Immune activation is required for NT-3-induced axonal plasticity in chronic spinal cord injury. Exp Neurol 2008; 209: 497–509.
Ramer LM, McPhail LT, Borisoff JF, Soril LJ, Kaan TK, Lee JH et al. Endogenous TrkB ligands suppress functional mechanosensory plasticity in the deafferented spinal cord. J Neurosci 2007; 27: 5812–5822.
Young W . Review of lithium effects on brain and blood. Cell Transplant 2009; 18: 951–975.
Phiel CJ, Klein PS . Molecular targets of lithium action. Annu Rev Pharmacol Toxicol 2001; 41: 789–813.
Etheridge SL, Spencer GJ, Heath DJ, Genever PG . Expression profiling and functional analysis of wnt signaling mechanisms in mesenchymal stem cells. Stem Cells 2004; 22: 849–860.
Xie N, Li H, Wei D, LeSage G, Chen L, Wang S et al. Glycogen synthase kinase-3 and p38 MAPK are required for opioid-induced microglia apoptosis. Neuropharmacology 2010; 59: 444–451.
Wen YR, Tan PH, Cheng JK, Liu YC, Ji RR . Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. J Formos Med Assoc 2011; 110: 487–494.
Parkitna JR, Obara I, Wawrzczak-Bargiela A, Makuch W, Przewlocka B, Przewlocki R . Effects of glycogen synthase kinase 3beta and cyclin-dependent kinase 5 inhibitors on morphine-induced analgesia and tolerance in rats. J Pharmacol Exp Ther 2006; 319: 832–839.
Gault D, Morel-Fatio M, Albert T, Fattal C . Chronic neuropathic pain of spinal cord injury: what is the effectiveness of psychocomportemental management? Ann Phys Rehabil Med 2009; 52: 167–172.
Grandjean EM, Aubry JM . Lithium: updated human knowledge using an evidence-based approach: Part I: Clinical efficacy in bipolar disorder. CNS Drugs 2009; 23: 225–240.
Fahy S, Lawlor BA . Lithium use in octogenarians. Int J Geriatr Psychiatry 2001; 16: 1000–1003.
Grandjean EM, Aubry JM . Lithium: updated human knowledge using an evidence-based approach. Part II: Clinical pharmacology and therapeutic monitoring. CNS Drugs 2009; 23: 331–349.
Yeung CK, Chan HH . Cutaneous adverse effects of lithium: epidemiology and management. Am J Clin Dermatol 2004; 5: 3–8.
Acknowledgements
We thank Dr Limin Liao, Dr Lan Sun, Dr Zhigang Chen, Dr Jimin Xu, Ms Mingqin Dong, Dr Xiandi Zhang and Dr Yukun Yang for significant contributions to this phase II clinical study. The study was supported by China Spinal Cord Injury Network Company Limited.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yang, M., Li, J., So, K. et al. Efficacy and safety of lithium carbonate treatment of chronic spinal cord injuries: a double-blind, randomized, placebo-controlled clinical trial. Spinal Cord 50, 141–146 (2012). https://doi.org/10.1038/sc.2011.126
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2011.126
Keywords
This article is cited by
-
Management of central neuropathic pain involves many drugs but few have proven efficacy
Drugs & Therapy Perspectives (2023)
-
Drug Repurposing for Spinal Cord Injury: Progress Towards Therapeutic Intervention for Primary Factors and Secondary Complications
Pharmaceutical Medicine (2023)
-
Coexistence of chronic hyperalgesia and multilevel neuroinflammatory responses after experimental SCI: a systematic approach to profiling neuropathic pain
Journal of Neuroinflammation (2022)
-
Central Neuropathic Pain Syndromes: Current and Emerging Pharmacological Strategies
CNS Drugs (2022)
-
The challenge of recruitment for neurotherapeutic clinical trials in spinal cord injury
Spinal Cord (2019)
