
Key Points
-
Positive and negative allosteric modulators (PAMs and NAMs) have now been identified for members of each major subfamily of G protein-coupled receptors (GPCRs). Multiple allosteric modulators have been optimized to have drug-like properties, and several allosteric modulators of class A and class C GPCRs are now advancing in preclinical and clinical development.
-
The crystal structures of GPCRs bound to allosteric modulators are providing important new insights into the structural determinants of allosteric-modulator action. This new structural information reveals interesting similarities between the binding pocket for allosteric modulators of class C GPCRs and the orthosteric binding pocket of class A GPCRs.
-
Potential advantages of allosteric modulators that were theoretical are now gaining direct experimental support. For instance, there are now clear examples of the major advantages of using GPCR PAMs as opposed to orthosteric agonists in maintaining spatial and temporal aspects of GPCR signalling.
-
Stimulus bias can complicate the optimization of GPCR allosteric modulators for disorders of the central nervous system, in which the most appropriate signalling pathways are not known. However, there are also examples where the optimization of allosteric modulators that confer stimulus bias to GPCR signalling may be important for developing optimal clinical-development candidates.
-
Examples are now beginning to emerge in which allosteric modulators can differentiate between homomeric and heteromeric forms of class C GPCRs. Allosteric modulators that can differentiate between homomeric and heteromeric GPCRs can have highly specific effects in certain brain circuits.
-
After a decade of focused efforts on the optimization of allosteric modulators of GPCRs as drug candidates, principles are now emerging that are important for the successful lead optimization of candidate allosteric modulators.
Abstract
Novel allosteric modulators of G protein-coupled receptors (GPCRs) are providing fundamental advances in the development of GPCR ligands with high subtype selectivity and novel modes of efficacy that have not been possible with traditional approaches. As new allosteric modulators are advancing as drug candidates, we are developing an increased understanding of the major advantages and broad range of activities that can be achieved with these agents through selective modulation of specific signalling pathways, differential effects on GPCR homodimers versus heterodimers, and other properties. This understanding creates exciting opportunities, as well as unique challenges, in the optimization of novel therapeutic agents for disorders of the central nervous system.
Similar content being viewed by others


Main
The G protein-coupled receptors (GPCRs), which are also known as seven-transmembrane receptors (7TMRs), are crucial for transducing signals from the extracellular environment into intracellular changes in cell function. GPCRs are activated by a diverse range of ligands and stimuli, including hormones, neurotransmitters, membrane lipids, ions, odorants, enzymes and photons of light, and have diverse roles in every cell and organ system. Human GPCRs are subdivided into four major classes (A, B, C and F) on the basis of their primary sequence homology1,2. Given the roles of GPCRs in cellular responses to extracellular ligands, it is not surprising that these receptors have been among the most successful of drug targets3.
Despite the proven success of GPCRs as drug targets, many intense efforts to develop selective ligands or drug candidates for GPCRs have failed. Thus, there is a major need to develop new approaches for the discovery of therapeutic agents that target this important class of receptors. Traditionally, drugs that target GPCRs bind to the same site as the endogenous agonist (referred to as the orthosteric site), to either activate or inhibit the receptor. Although this is a strategy well suited for some receptors, many GPCRs respond to stimuli such as light, protons, divalent cations, highly charged nucleotides, peptides and proteins; in many of these cases, these endogenous ligands cannot be easily modified to retain drug-like properties. For example, many amino acids and peptides cannot cross the blood–brain barrier, meaning that alternative ligands must be developed to modulate certain targets in the central nervous system (CNS). In addition, some GPCRs are activated by enzymatic cleavage. Alternative strategies for modulation may provide advantages in the development of therapeutic agents that target some receptors. Furthermore, as orthosteric binding sites are often highly conserved, it is difficult to achieve high selectivity for individual GPCR subtypes. The need to develop new approaches for targeting GPCRs is especially urgent for neurological and psychiatric diseases, which have been especially challenging to address therapeutically and have historically had a very high attrition rate4.
Allosteric modulators of GPCRs
Interestingly, CNS drug discovery efforts to target ligand-gated ion channel neurotransmitter receptors have focused primarily on developing ligands that interact with allosteric sites that are topographically distinct from the orthosteric neurotransmitter site5,6. By binding to allosteric sites, such ligands can act as either positive allosteric modulators (PAMs) or negative allosteric modulators (NAMs) to potentiate or inhibit activation of the receptor by the endogenous agonist, respectively. Selective PAMs of GABAA (γ-aminobutyric acid type A) receptors, such as benzodiazepines and barbiturates, were among the earliest-approved drugs for the treatment of CNS disorders7. These allosteric modulators can provide many potential advantages over traditional agonists, including the requirement for the synaptic release of the neurotransmitter and the activation of the receptor by the endogenous agonist. However, optimization of allosteric modulators for GPCRs did not become practicable until the late 1990s and early 2000s, when the advent of high-throughput functional screening technologies enabled the study of the functional effects of small molecules on GPCR signalling that were independent of the effects on orthosteric ligand binding. Over the past decade, the discovery and understanding of allosteric modulators have gained tremendous momentum, and GPCR allosteric modulators are now marketed as therapeutic agents for the treatment of hyperparathyroidism8,9 and HIV infections10.
To date, no GPCR allosteric modulators have been approved for the treatment of psychiatric and neurological disorders. However, multiple allosteric modulators have entered clinical development (Table 1). Phase II clinical trials have now demonstrated promising effects of metabotropic glutamate receptor 5 (mGluR5) NAMs in anxiety and affective disorders, Parkinson's disease and fragile X syndrome11,12. More recently, a novel mGluR2 PAM entered Phase II testing for schizophrenia and anxious depression13,14, and Merck has announced the initiation of a Phase II trial of a PAM that targets the M1 receptor — a subtype of muscarinic acetylcholine receptors (mAChRs) — in patients with Alzheimer's disease (ClinicalTrials.gov identifier: NCT01852110). Other allosteric modulators are now in late discovery or preclinical development, including mGluR5 PAMs for schizophrenia15; mGluR4 PAMs for Parkinson's disease16; and M4-receptor PAMs for the treatment of psychosis associated with schizophrenia and psychosis associated with Alzheimer's disease17 (Table 1). Results from early optimization and animal studies are now emerging for allosteric modulators of a range of other CNS GPCRs from families A (Supplementary information S1 (table)), B (Supplementary information S2 (table)) and C (Supplementary information S3 (table)) and are indicative of a healthy pipeline of novel allosteric modulators that hold promise for the treatment of multiple neurological and psychiatric disorders.
Allosteric modulator pharmacology
PAMs, NAMs and NALs. GPCRs are dynamic proteins that spontaneously transition between multiple functionally distinct conformational states18,19. These include states with varying affinities for orthosteric ligands, as well as different interactions with G proteins and other effector or signalling molecules. PAMs and NAMs are thought to stabilize receptors in specific conformational states that increase or decrease the functional response to orthosteric agonists, respectively. Both PAMs and NAMs can modulate the affinity of the orthosteric binding pocket for an orthosteric ligand or affect the intrinsic efficacy of an orthosteric agonist to engage downstream signalling mechanisms19. Allosteric modulators within a structural class can each have any one of a broad range of effects on GPCRs, including positive or negative modulation, or even inverse agonism (where the constitutive activity of a receptor is reduced by ligand binding). Within this continuum, neutral allosteric ligands (NALs) can bind to allosteric sites, but have no effect on receptor responses to orthosteric ligands; however, these agents compete with PAMs and NAMs for binding to allosteric sites and block their actions19,20,21. Although they are not pursued as drug candidates themselves, NALs provide important tools to validate binding sites and could theoretically serve as candidates for radiolabelling or PET-(positron emission tomography)-ligand development if they possess sufficiently high affinity.
The diverse effects of allosteric ligands on receptor behaviour have been outlined in detail in multiple excellent reviews and can be described and quantified by equations that are based on various ternary-complex mass-action models. These equations take the interactions among the receptor, orthosteric ligands, allosteric ligands and effector proteins into account and provide a computational approach to define the magnitude and direction of an allosteric effect, using one or more cooperativity factors18,22. More recently, an operational model of allosterism that describes the allosteric modulation of both affinity and efficacy was introduced; this model also incorporates the ability of a compound to induce allosteric agonism23,24,25. Briefly, in the operational model, modulation of binding affinity is governed by a cooperativity factor, which is denoted as α, and allosteric modulation of efficacy is incorporated by the introduction of the parameter β. In addition, a composite cooperativity parameter (known as logαβ) can be used to incorporate the modulation of both affinity and efficacy. As some PAMs also have intrinsic efficacy in the absence of an orthosteric agonist, the parameters τΑ and τB have also been incorporated to account for the intrinsic efficacy of the orthosteric and allosteric ligands, respectively.
This operational model of allosterism has been highly useful in quantifying different aspects of allosteric modulator function and can provide valuable information about cooperativity factors and the structure–activity relationships (SARs) of predicted affinities when conducting a lead-optimization programme. To test the validity of the model, Gregory et al.26 recently took advantage of the radioligands and the range of allosteric modulators for mGluR5. The study confirmed that for mGluR5 PAMs and NAMs, with diverse chemical scaffolds and varying degrees of cooperativity, affinity estimates that were derived from functional assays that used the operational model fitted well with affinities that were measured in radioligand-binding experiments. It should be noted, however, that the functional estimates of affinity may change depending on the signalling pathway being measured. As such, estimates of functional affinity may actually be more relevant than the binding affinity when considering a particular signalling cascade. This point is also important if a correlation between the inhibition constants (Kis) that are predicted using traditional binding assays do not align with those predicted by functional affinity assessments.
Allosteric agonists and ago-PAMs. As mentioned above, in addition to potentiating responses to orthosteric agonists, GPCR PAMs can have intrinsic efficacy and activate the receptor in the absence of an orthosteric agonist. PAMs that also possess intrinsic allosteric agonist activity in a given functional assay are referred to as ago-PAMs; however, the presence or absence of ago-PAM activity may depend on the assay system that is used for assessment, the level of receptor expression or the efficiency of receptor coupling with a given second-messenger system27,28. Thus, β and τB values for an allosteric ligand are determined not only by the individual ligands but also by the biological assay system that is being investigated. For this reason, when evaluating effects of a given PAM in systems with low stimulus–response coupling efficiency or low levels of receptor expression, τΒ values may approach 0 and ago-PAM activity may not be discernible, despite robust potentiation of orthosteric agonist responses. However, when measuring responses in cells with high receptor expression and/or coupling efficiency, the same PAM may display high τΒ values and marked ago-PAM activity in a given assay27,28,29,30.
This discrepancy was recently demonstrated for a series of mGluR5 PAMs using a cell line in which mGluR5 was expressed under the control of an inducible promoter28. In cells in which high levels of mGluR5 expression were induced, multiple mGluR5 PAMs showed robust ago-PAM activity in inducing calcium-mobilization responses, but in cell lines that expressed only low levels of mGluR5, these mGluR5 PAMs exhibited no discernable allosteric agonist activity. However, the efficacies and potencies of these same PAMs in potentiating the response to glutamate were not notably affected by changes in mGluR5 expression28. Interestingly, the compounds that displayed ago-PAM activity in calcium assays in cells that overexpressed mGluR5 — but not in cells in which lower levels of mGluR5 were expressed — behaved as 'pure' PAMs with no discernible agonist activity when multiple responses were measured in native systems using electrophysiological readouts. However, in a subsequent study29, mGluR5 PAMs were intentionally optimized to induce robust calcium responses in the absence of exogenously applied glutamate, even in cells with low levels of mGluR5 expression. These mGluR5 ago-PAMs exhibited strong agonist activity in multiple native systems, providing a direct demonstration of the effect of receptor expression on τΒ, and of the potential context dependence of responses to specific PAMs. It should be noted, however, that in addition to modulation by receptor density, alterations in τΑ and τB are also dependent on signalling pathways, and τ values may not change equally for different pathways. Recent studies with mGluR5 PAMs provide direct evidence that PAMs for this receptor subtype maintain activity dependence in certain CNS circuits31.
In addition to ago-PAMs, GPCR ligands can bind to sites that are distinct from the orthosteric site and have direct agonist activity without potentiating responses to orthosteric agonists. Of such ligands, allosteric agonists of mAChRs have been the most widely studied. Early studies suggested that M1-receptor allosteric agonists may have major advantages over orthosteric M1-receptor agonists as they are comparatively more selective for the M1-subtype receptors32,33,34,35,36; however, most M1-receptor allosteric agonists are so-called 'bitopic ligands' that bind simultaneously both to an allosteric site and to the orthosteric site37,38,39 and require engagement of the orthosteric site for their activity. In these cases, the compounds bind to the orthosteric sites across all mAChR subtypes, but exhibit functional selectivity for certain subtypes38. Most M1-receptor bitopic agonists tend to act as weak partial agonists at M1 receptors and exhibit only weak agonist activity in systems where M1 receptors are expressed with low receptor reserve. Owing to this weak partial agonist activity, allosteric agonists can suffer from the same problems as do traditional M1-receptor orthosteric agonists and it can be difficult to achieve high selectivity38. Although it is not yet clear whether the same loss of selectivity would be observed with allosteric agonists of other receptor subtypes, these findings present major issues for the optimization of M1-receptor bitopic agonists as drug candidates owing to dose-limiting off-target activity; however, this problem could be avoided by M1-receptor PAMs or ago-PAMs, as long as they do not engage the acetylcholine (ACh) binding site.
Partial antagonists. The discovery of GPCR NAMs that act by modulating efficacy (β) raises the exciting possibility that NAMs that modulate β with low levels of negative cooperativity could act as 'partial antagonists' that only partially reduce agonist responses when the allosteric site is fully occupied40. It has been postulated that because 'partial' NAMs exhibit limited negative cooperativity in blocking orthosteric agonist responses, they might reduce the adverse-effect profiles that may be associated with complete blockade of some GPCRs using full NAMs or inverse agonists. However, this potential advantage remains theoretical as, to date, partial NAMs have not been optimized for use in in vivo studies. The same potential advantage exists for PAMs that show limited levels of positive cooperativity; with these ligands, a saturation of effect is postulated to provide safety in the event of an overdose, as exemplified by benzodiazepine PAMs of GABAA receptors7. Several examples of partial antagonists that act exclusively by modulating efficacy have now been identified21,41. However — as with orthosteric partial agonists, allosteric agonists and ago-PAMs — partial antagonist activity can depend on the level of receptor expression or coupling efficiency. For example, Gregory et al.26 reported that three low-cooperativity mGluR5 NAMs behaved as partial antagonists in cells that expressed low levels of mGluR5, but induced greater decreases in glutamate responses in cells that expressed high levels of mGluR5. Thus, it will be important to consider the impact of receptor expression and coupling efficiency on different native-system responses when evaluating compounds that have been optimized to induce low levels of cooperativity with the orthosteric agonist.
Structural determinants of allosteric modulation
Revealing the structural determinants of the interactions between allosteric modulators and GPCRs will be crucial for the understanding of the mode of action of allosteric ligands and to allow for structure-based drug discovery. Early modelling and mutagenesis studies provided insights into the NAM binding pocket of mGluR1 (Refs 42,43), and this was followed by extensive mutagenesis and computational docking studies using models of mGluRs that were based on insights from crystal structures of class A GPCRs44,45. These studies identified an allosteric binding pocket in the seven-transmembrane domain (TMD) that is a common binding pocket for a range of mGluR allosteric modulators and pinpointed specific residues that are crucial for the activity of PAMs, NAMs and NALs. Similar mutational analyses and modelling studies suggest that allosteric modulators of class A GPCRs can also bind to the TMD but, in addition, engage the extracellular loops (ECLs) of the receptor46. Specifically, nuclear magnetic resonance (NMR) spectroscopic studies of the β2-adrenergic receptor suggest that orthosteric agonists induce a reshaping of the extracellular surface — including a salt bridge linking ECL2 and ECL3 — that may reveal highly specific binding sites for allosteric modulators47.
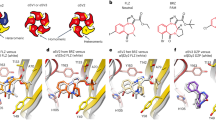
Over the past year, the first two co-crystal structures of allosteric modulators in complex with the TMDs of class A and class C GPCRs have been reported48,49. Furthermore, the co-crystal structure of an allosteric antagonist in complex with the TMD of a class B GPCR has also been reviewed, thus completing the structures of the three main GPCR groups50. Insights from these crystal structures fit well with existing models and greatly expand our understanding of GCPR structure–function relationships (Fig. 1). Notably, the increasing number of GPCR crystal structures will allow comparative modelling and docking studies51, as well as molecular-dynamics simulations52, to contribute more substantially to the understanding of the structural basis of allosteric modulation.

a | Interaction of a class A GPCR (G protein-coupled receptor) — the M2 subtype muscarinic acetylcholine receptor (Protein Data Bank (PDB) identifier: 4MGT) — in complex with the positive allosteric modulator (PAM) LY2119620 (pink) and the orthosteric ligand iperoxo (white). Y177 and W422 engage the PAM via π–π interactions. Y426 separates the allosteric ligand binding site from the orthosteric ligand binding site. b | Interaction of the class B corticotropin-releasing factor receptor type 1 (CRF1 receptor) with the allosteric antagonist CP-376395 (pink; PDB 4K5Y). The binding pocket in the intracellular half of the seven-transmembrane domain is dominated by hydrophobic interactions. The conserved N283 forms a crucial hydrogen bond with the ligand. c | Interaction of the class C metabotropic glutamate receptor type 1 (PDB 4OR2) with the negative allosteric modulator FITM (pink). The binding pocket overlaps with the orthosteric ligand binding site in class A GPCRs. It is largely hydrophobic, with T815 contributing an important hydrogen bond to the ligand.
Allosteric modulation of the M2 receptor. The first study reports the structure of the class A mAChR, M2, in complex with the PAM LY2119620 (Ref. 48) and reveals a complex that includes both the PAM and an orthosteric agonist (iperoxo). LY2119620 enhances the affinity of the M2 receptor for iperoxo and behaves as an ago-PAM in GTP-γS-binding and extracellular signal-regulated kinase 1 (ERK1)- or ERK2-phosphorylation assays. Consistent with previous homology modelling and docking studies that implicate a role for ECL2 (Refs 39,53), the co-crystal structure reveals that the allosteric modulator is positioned above the orthosteric ligand and interacts with the extracellular vestibule by forming π–π interactions with a tyrosine residue in the ECL2 and a tryptophan residue at the top of transmembrane-spanning domain 7 (TM7) (Fig. 1a). The ligand is further positioned by two hydrogen bonds and a salt bridge. LY2119620 does not directly interact with iperoxo, but the two binding sites are separated only by Tyr4267.39, which interacts with both ligands. The structure of the M2 receptor TMD is largely independent of the presence of LY2119620, suggesting that the allosteric binding pocket is pre-formed when iperoxo binds. The largest change that is observed in the presence of LY2119620 is a re-orientation of the Trp4227.35 side chain to form the π–π interaction with LY2119620 and a slight contraction of the allosteric binding site. This reduction is small, however, compared with the marked contraction of the allosteric site that is observed upon the binding of the orthosteric agonist. The tightening of the allosteric site upon agonist binding is a direct effect of the inward motion of TM6, which contacts the LY2119620, iperoxo and the G protein. These observations are consistent with the notion that LY2119620 enhances the binding affinity of the agonist by slowing its dissociation; in addition, these results confirm that LY2119620 has poor agonist activity and affinity for the inactive conformation of the receptor.
Allosteric antagonist bound to the CRF1 receptor. No co-crystal structure of a class B GPCR with a PAM has been revealed. However, the corticotropin-releasing factor receptor 1 (CRF1 receptor) has been co-crystallized with a NAM, CP-376395 (Ref. 50). CRF1 receptors mediate the response to stress and have been considered to be a drug target for depression and anxiety. CP-376395 binds in a hydrophobic pocket in the cytoplasmic half of the receptor, about 18 Å below the orthosteric binding site of the peptide agonist or the orthosteric binding sites of class A GPCRs (Fig. 1b). The most prominent polar contact is a hydrogen bond of the pyridine nitrogen with the highly conserved Asn2835.50. The ligand is separated from the orthosteric site by a layer of residues that include Arg1652.60, His1993.40, Met2765.43 and Gln3557.49. It is proposed that, when in this location, the ligand tethers TM6 to TM3 and TM5, thereby locking the receptor in an inactive conformation. This proposition is supported by the fact that CP-376395 acts as an inverse agonist in reducing basal signalling. A series of compounds with a range of allosteric effects are expected to bind in this region of the receptor54.
Allosteric modulation of mGluR1. The second direct measure of an interaction of an allosteric modulator with the TMD has been conducted on the class C GPCR mGluR1 (Ref. 49). In contrast to the M2 receptor, mGluR1 is activated by glutamate binding to a large extracellular domain called the Venus-flytrap domain (VFD)55. Conformational changes of the VFD are transduced via a cysteine-rich domain (CRD) to the TMD. The TMD was co-crystallized with a NAM called FITM56, which binds to a pocket that is formed by the TMD and lies just above the membrane centre (Fig. 1c). This pocket is directly analogous to the pocket that many orthosteric ligands of class A GPCRs, such as iperoxo and ACh, leverage in M2 receptors. The similarity between the allosteric site on mGluR1 and the orthosteric site on class A GPCRs is especially striking in light of the absence of sequence homology between these two GPCR subfamilies. Indeed, this site similarity further reinforces the remarkable similarities in the function and three-dimensional topology of the crystal structures of members of the different GPCR subfamilies that are observed, despite the relatively low sequence homology between the subfamilies57.
Although the allosteric binding pocket in mGluR1 shares common features with the orthosteric binding pocket in class A GPCRs, there are also important differences. The TMD of mGluR1 is more compact than those of class A GPCRs49, thus limiting space and access to the NAM binding site. The mGluR1 binding pocket involves side chains from residues in TMD helices 2, 3, 5, 6 and 7, as well as ECL2. Consistent with the compact TMD, the pocket is elongated and narrow. Ligand–receptor interactions are dominated by tightly fitted hydrophobic interactions; only one hydrogen bond to Thr8157.38 is observed. In particular, the intracellular site of the TMD adopts an inactive conformation which is stabilized by a salt bridge between Lys6783.46 and Glu7836.33 in an analogy to the 'ionic lock': a salt bridge that is observed between Arg3.50 and the acidic residue in the 6.30 position of class A GPCRs. This conformation of the TMD is consistent with FITM acting as a NAM and suggests that FITM stabilizes the TMD in this locked conformation. It is interesting to note that previous mutagenesis, pharmacological and radioligand-binding studies have indicated that multiple mGluR1 NAMs bind to a common site that is not shared by known mGluR1 PAMs58,59. This is in contrast to mGluR5, where NAMs, PAMs, NALs and ago-PAMs can all bind to a single site that is thought to be homologous with the most prominent allosteric site for NAMs on mGluR1 (Refs 26,44,58,60). As the structure of mGluR1 in complex with a NAM has now been elucidated, it will be important to determine whether mGluR1 PAMs interact with the same site as that occupied by FITM. It will also be interesting to see whether interaction with a PAM stabilizes a receptor conformation that prevents the formation of the salt bridge between Lys6783.46 and Glu7836.33, thereby disrupting the ionic lock and pushing the receptor into an active conformation. Such studies — using a known structure — will hopefully aid in translation to other mGluRs, such as mGluR2, mGluR4 and mGluR5, that are primary drug targets for selective PAMs.
Optimizing synthetic GPCR allosteric modulators
The complexity of the actions of allosteric modulators raises unique challenges for lead-optimization efforts that are aimed at advancing these agents as drug candidates. As discussed below, recent studies are now providing direct evidence that GPCR allosteric modulators can provide many of the predicted advantages over orthosteric ligands, and an understanding of approaches to address the complexities of allosteric modulators in lead-optimization efforts is beginning to emerge.
Selectivity. A primary driver for the early drug discovery efforts that were focused on allosteric modulators for GPCRs was the hope of developing greater subtype selectivity within the receptor classes that had previously proved to be intractable using traditional orthosteric approaches. For instance, multiple efforts to develop highly selective glutamate binding-site agonists and antagonists for any one of the eight individual subtypes of mGluRs (mGluR1–mGluR8) had failed, probably owing to the high conservation of the orthosteric glutamate binding site across the subtypes. However, the discoveries of the mGluR1 NAM CPCCOEt61,62 and the mGluR5 NAM SIB-1757 (Ref. 63) provided the first highly selective antagonists for any individual mGluR subtype. These developments stimulated intense efforts that have yielded highly selective NAMs and PAMs for most of the eight mGluR subtypes20,27,41,58,64,65,66,67,68,69,70,71. Another major validation for achieving high subtype selectivity came with the discovery of highly selective allosteric agonists and PAMs for several individual subtypes of mAChRs, including M1 (Refs 72,73,74), M4 (Refs 75,76) and M5 (Refs 71,77). This was especially encouraging in light of the tremendous efforts to develop selective agonists of individual mAChR subtypes in the 1990s, all of which had failed to achieve high subtype selectivity17,35. It is especially striking that high subtype selectivity appears to be the norm, rather than the exception, for many GPCR allosteric modulators. Many allosteric modulators that have been identified via screening display subtype selectivity without further optimization and, in other cases, medicinal-chemistry efforts have been highly successful in achieving subtype selectivity77,78. Furthermore, the broad profiling of a limited number of GPCR allosteric modulators for their modulatory effects on other GPCRs has revealed remarkable selectivity for the intended GPCR relative to a broad range of other receptors27,34,35,41,66,68,69,70,71,79. However, it should be noted that selectivity can be dependent on the assay used for assessment, as well as the probe compound that is used in conjunction with the allosteric modulator. For example, the M4-receptor PAM LY2033298 binds to the M2 receptor and exhibits differential cooperativity, both positive and negative, with distinct orthosteric agonists in binding assays, as well as in functional assays such as ERK phosphorylation and GTP-γS binding80. Additionally, the use of receptors from different species in the assessment of allosteric ligand activity may also reveal differences in cooperativity, probe dependence or ago-PAM activity, depending on the pathway and ligand that are used81. For instance, several mGluR1 NAMs have been optimized — using cell lines that expressed either rat or human mGluR1 — that later proved to be relatively inactive at mGluR1 in the other species82. Likewise, LY2033298, an M4-receptor PAM75, is highly selective for human M4 receptor over rodent M4 receptor, in terms of both affinity and cooperativity81. Furthermore, many receptors may respond to more than one endogenous ligand; therefore, the activity of a modulator should be assessed in the presence of different endogenous ligands. The above points require that the activity of a compound is carefully profiled in multiple assays, across species and against additional ligands, as these points are key to the interpretation of in vivo pharmacology studies.
Maintenance of spatial and temporal aspects of GPCR signalling and the impact of ago-PAM activity. Another potential advantage of GPCR PAMs is that these compounds may preserve the normal function of endogenous neurotransmitters and enhance neurotransmitter action in a more physiologically appropriate manner. Recent studies with mGluR5 PAMs provide direct evidence that PAMs for this receptor subtype maintain activity dependence in certain CNS circuits31. These studies are described in Box 1. The striking negative impact of the loss of this advantage with compounds that possess mGluR6 ago-PAM activity has proven to be crucial for the optimization of mGluR5 PAMs as potential therapeutic agents. Although the elimination of allosteric agonist activity is not the only factor that is important for developing safe and effective mGluR5 PAMs, efforts to optimize mGluR5 PAMs for clinical development must strictly avoid ago-PAM activity. We have monitored for agonist activity, using calcium-mobilization assays in cell lines and astrocytes as well as using brain-slice electrophysiology, to profile mGluR5 responses in the hippocampus, prefrontal cortex and striatum. As mentioned above, the ability to detect the ago-PAM activity of mGluR5 PAMs depends in part on receptor expression levels and the efficiency of receptor–response coupling28. Thus, it is important to ensure that any potential for ago-PAM activity is assessed at different levels of receptor expression and in native systems29,83.
Although it appears to be important to avoid ago-PAM activity for mGluR5 PAMs, the preferred profile must be carefully considered and empirically determined for each programme or target. If tonic levels of the endogenous agonist are low, there may be a need for high cooperativity and ago-PAM activity to achieve maximal efficacy. For instance, ago-PAM activity could be beneficial in efforts to optimize M1-receptor PAMs or M4-receptor PAMs for the treatment of Alzheimer's disease, which is associated with a loss of cholinergic neurons early in the course of the disease84, or for mGluR4 PAMs for the treatment of Parkinson's disease, in which mGluR4 agonists have shown robust efficacy85.
Stimulus bias. GPCRs do not only exist in simple inactive or active conformations, but activate a broad range of signalling pathways, including both G protein-dependent and G protein-independent signalling. Agonists can stabilize multiple active states that can differentially engage different signalling pathways, thereby biasing the receptor in favour of the activation of specific signalling pathways86,87. In addition, allosteric modulators have the potential to selectively modulate the ability of agonists to stabilize specific active conformations of the receptor and thereby introduce a stimulus bias that differentially alters the effects on specific signalling pathways of the endogenous agonist.
Over the past several years, multiple GPCR PAMs have been shown to differentially alter signalling pathways that are activated by endogenous neurotransmitters. For instance, VU0029767 and VU0090157 are selective M1-receptor PAMs, both of which induce comparable potentiation of ACh-induced calcium mobilization in M1-receptor-positive cells. Furthermore, VU0090157 potentiates the ACh-induced activation of phospholipase D (PLD) — similarly to enhancing calcium mobilization — whereas VU0029767 has no effect on M1-receptor-mediated activation of PLD in the same cell background that was used in the calcium-mobilization studies73. The M1-receptor-mediated calcium mobilization and activation of phospholipase C involves signalling through Gαq, whereas PLD typically involves the activation of Gα12 or small G proteins such as the RAS-related protein RRAS88. Thus, it is possible that VU0090157 potentiates the ability of ACh to stabilize conformations that activate both signalling pathways, whereas VU0029767 may selectively potentiate the ability of ACh to stabilize a conformation of the M1 receptor that couples to Gαq but is unable to form productive signalling complexes with Gα12 or small G proteins. Although the precise roles of these signalling pathways in different physiological and behavioural responses to M1-receptor activation are not known, closely related M1-receptor PAMs that have such striking differences in their effects on M1-receptor signalling could have fundamentally different effects on animal behaviour and could display distinctions in therapeutic efficacy or adverse-effect liability. Similar examples of biased signalling in cell lines have now been observed for PAMs that act at multiple GPCR subtypes, including M4 receptors89, mGluRs90,91, calcium-sensing receptors92,93 and cannabinoid receptors94. In addition, there are now multiple examples of NAMs that selectively inhibit specific signalling pathways that are activated by the endogenous agonist. This includes biased or functionally selective NAMs for mGluR7 (Ref. 95), prostaglandin D2 receptors96 and substance K (also known as neurokinin 2) receptors97. The unique potential of GPCR NAMs to selectively inhibit agonist effects on specific signalling pathways is not shared by orthosteric antagonists and provides an exciting opportunity to develop biased NAMs that target the pathways that are most crucial for achieving a therapeutic effect.
Most examples of biased signalling with GPCR modulators have been observed in cell lines that express recombinant receptors. However, recent studies reveal that mGluR5 PAMs can display stimulus bias in CNS preparations, and this directly affects the functional response to different mGluR5 PAMs. As discussed above, mGluR5 PAMs have been a major focus for the development of novel agents for reducing positive symptoms and enhancing cognitive function in patients with schizophrenia. One possible effect of mGluR5 activation in the CNS that could be important for the therapeutic efficacy of mGluR5 PAMs is the potentiation of currents through the NMDA (N-methyl-d-aspartate) subtype of ionotropic glutamate receptors and an NMDA-receptor-dependent form of synaptic plasticity that is termed long-term potentiation (LTP). However, increased activation of NMDA receptors can also lead to excitotoxicity and cell death, and it is possible that the mGluR5-mediated potentiation of NMDA-receptor currents contributes to the cell death and toxicity in the CNS that has been observed with some mGluR5 PAMs. Interestingly, a novel mGluR5 PAM known as NCFP potentiates multiple mGluR5-mediated responses in both recombinant and native systems, but does not potentiate mGluR5-mediated enhancement of hippocampal LTP98, suggesting a strong bias or context-dependent pharmacology that could have an impact on the in vivo effects of mGluR5 PAMs. More recently, VU0409551 was optimized as a novel mGluR5 PAM that potentiates multiple responses to mGluR5 activation but does not potentiate hippocampal LTP or mGluR5 coupling to the modulation of NMDA-receptor currents in hippocampal neurons99. Interestingly, VU0409551 has robust antipsychotic-like effects and enhances hippocampal-dependent cognitive function in multiple animal models. VU0409551 does not induce adverse effects such as seizures, and does not induce cell death in the CNS at doses tenfold greater than those needed for efficacy in rodent models. These studies suggest that biased mGluR5 PAMs that retain efficacy in rodent models, despite not potentiating NMDA-receptor currents, can be optimized. Such biased mGluR5 PAMs could provide an advantage in avoiding NMDA-receptor-mediated adverse-effect liability and CNS toxicity.
Although the ability of allosteric modulators to confer or introduce stimulus bias presents potential opportunities to develop more highly selective drug candidates, the potential for stimulus bias also introduces complexities for the optimization of GPCR allosteric modulators as therapeutic agents. Signalling in the CNS is complex and a given GPCR can couple to different signalling partners in different cell populations. In most cases, we do not have a clear understanding of the specific signalling pathways that are crucial for therapeutic efficacy or that mediate adverse effects. However, it is generally agreed that understanding signalling bias may ultimately become crucial for reducing failures in predicting drug efficacy; definitively linking a given pathway to a desired pharmacological response will certainly be one of the horizons of GCPR drug discovery.
In the absence of this understanding, however, perhaps a more practical approach is to proceed in parallel, by optimizing compounds that have no apparent stimulus bias. Inadvertent optimization of allosteric modulators that induce an unrecognized stimulus bias could lead to the discovery of compounds that are potent and have high efficacy in the cell-based assay used to drive chemical optimization, but display an unexplained lack of efficacy in native systems, in vivo models or clinical studies. For instance, early optimization of a series of mGluR7 NAMs using a calcium-fluorescence assay yielded highly selective NAMs with nanomolar potency, that nonetheless failed to block functional responses to mGluR7 activation in the hippocampal formation95. This context-dependent NAM activity was initially missed when a single assay for mGluR7 function was used, but became evident when the effects of these mGluR7 NAMs were investigated in other cell-based assays and native tissues. Although it is not practical to incorporate a broad range of functional assays in a rapidly advancing chemistry programme, the potential for unknowingly optimizing biased modulators can be largely mitigated by establishing multiple assays of receptor signalling in order to choose initial hits and scaffolds that are devoid of biased signalling. These assays can then be used at key points during the optimization effort, especially following major structural changes to a chemical scaffold, to ensure that the chemistry does not inadvertently introduce bias into a lead series. Although the assays that are used may not entirely reflect the unknown signalling pathways in the CNS that are required for efficacy, this strategy favours the optimization of compounds that potentiate a range of pathways that are engaged by the target receptor. In cases where the optimization of biased compounds is desired, it is crucial to include assays to detect bias in the CNS circuits that are being targeted, as — depending on the specific cell population — a given receptor may engage a given signalling pathway via multiple mechanisms.
Modulation of affinity versus efficacy. In addition to the potential for inadvertently optimizing compounds that differentially modulate the coupling of a receptor to distinct signalling pathways, it is also possible to optimize allosteric modulators that have effects on agonist affinity that do not predict functional changes in receptor activity. Because allosteric modulators can modulate both agonist affinity and coupling efficiency, it is possible for an allosteric modulator to have differential effects on the affinity versus the efficacy of a given orthosteric agonist. A prominent example of this is the case of the cannabinoid type 1 (CB1) receptor allosteric modulator Org27569, which is an allosteric enhancer of agonist binding, but an allosteric inhibitor of CB1 receptor coupling efficiency, and functionally acts as a NAM100. Another CB1 receptor modulator, PSNCBAM-1, has PAM-like effects on α, but functions as a NAM owing to negative cooperativity with respect to the modulation of β101. Thus, the use of measures of potentiation of agonist binding to drive SAR optimization could yield compounds that display orderly SARs for PAM activity when assessed with agonist binding but that act as functional NAMs or have a range of functional actions, including NAM-like, NAL-like or PAM-like activities, that bear no relationship to the effects on agonist affinity. Because of this potential, it is crucial to use functional measures of allosteric modulator activity to drive the SAR in lead-optimization efforts.
Differential effects of allosteric modulators on GPCR homodimers versus heterodimers. Numerous GPCRs have been reported to form functional dimers in vitro. Among the class A GPCRs, related members of the same subclass, such as the melatonin receptors MT1 and MT2 (Refs 102,103), or members from different families, such as the adenosine A1 and dopamine D1 receptors104, can form heterodimers in cell lines. Several other class A GPCR pairs also interact in expression systems105,106,107 and evidence suggests that heteromers and homomers of class A GPCRs can exhibit distinct orthosteric pharmacological profiles103 and signalling properties108,109 and induce different cell-trafficking characteristics110,111. However, to date, most studies of class A GPCR heteromers have been performed in cell lines, and so the extent to which class A GPCRs function as heteromers in native systems is not yet clear105. Also, although multiple studies reveal different effects of orthosteric ligands at homomeric versus heteromeric forms of class A GPCRs, the effect of the heteromeric assembly of the other classes of GPCRs on the responses to allosteric modulators has not been extensively studied. There are examples of heteromeric interactions between class B GPCRs (reviewed in Ref. 112); in some cases these have been identified as hetero-oligomerizations rather than dimerizations. Many class B GPCRs, such as the calcitonin gene-related peptide (CGRP) receptor and the secretin receptors, form complexes with accessory proteins that are known as receptor activity-modifying proteins (RAMPs). Although class B GPCRs do not truly heterodimerize, these important RAMP interactions can control class B GPCR trafficking and pharmacology. In the case of CGRP receptors, co-assembly with distinct RAMPs creates pharmacologically distinct receptors that respond to different endogenous peptides or small-molecule ligands112. The clinically used (non-allosteric) antagonists olcegepant and telcagepant interact with the CGRP receptor when it is bound to RAMP1, but not RAMP2 (Ref. 112).
In contrast to the class A GPCRs, some class C GPCRs, including the GABAB receptors, require assembly into a heteromeric form for normal function113,114, whereas other class C GPCRs, such as the mGluRs, can function as constitutive homodimers115. However, recent fluorescence resonance energy transfer (FRET)-based studies demonstrated that specific subgroups of mGluRs (groups 1, 2 and 3) can also interact as heterodimers in cell lines116. These findings were extended to evaluate the effects of allosteric modulators on putative heterodimers after co-expression of mGluR2 and mGluR4 in rat superior cervical ganglion cells117. These studies confirmed that mGluR2 and mGluR4 can interact in this cellular background and suggested that the occupation of only one orthosteric site (that is, either the mGluR2 or mGluR4 protomer of the dimer) was insufficient to activate the dimer, but that co-application of orthosteric agonists that were selective for mGluR2 and mGluR4 induced functional heterodimer activity. Interestingly, an mGluR2 NAM, Ro 64-5229, was ineffective in blocking glutamate responses in cells that expressed both mGluR2 and mGluR4. This result had been predicted from previous work using receptor mutagenesis that suggested that both protomers of an mGluR must be occupied by a NAM to block signalling from the extracellular domain118,119. Intriguingly, however, these experiments presented the first evidence that the mGluR2 PAM BINA and the mGluR4 PAMs PHCCC and VU0361737 do not appear to potentiate agonist activation of mGluR2–mGluR4 heterodimers. In addition, simultaneous application of an mGluR2 PAM and an mGluR4 PAM did not restore potentiation, suggesting that the mGluR2–mGluR4 heterodimer has a unique pharmacological profile in its response to allosteric modulators that is clearly distinct from homomeric mGluR2 or mGluR4 receptors.
More recently, these studies were extended in cell lines and native tissues in order to further our understanding of the unique profile of allosteric modulators on mGluR2–mGluR4 heterodimers and to provide evidence that this heterodimer may have a unique role that is distinct from that of homomeric mGluR2 and mGluR4 receptors in regulating activity in certain brain circuits120. Studies in human embryonic kidney 293 (HEK293) cells confirmed that mGluR4 PAMs, such as PHCCC and 4-PAM2, induced robust potentiation of mGluR4 responses when the mGluR4 was expressed alone, but were inactive as potentiators on the mGluR2–mGluR4 heterodimer120 (Fig. 2). By contrast, other PAMs, represented by VU0155041 and Lu AF29134 (Ref. 69), exhibited robust or even enhanced PAM activity on the mGluR2–mGluR4 heterodimer. Interestingly, PHCCC and 4-PAM2 are thought to bind to an allosteric site in the TMD of mGluR4 that is distinct from the TMD site that is occupied by VU0155041 and Lu AF21934 (Ref. 121). Modelling of the experimental results using the operational model of allosterism suggested that PHCCC and 4-PAM2 exhibited the same affinity regardless of whether homodimers or heterodimers were present, but showed a loss of cooperativity with orthosteric agonists. By contrast, VU0155041 and Lu AF21934 actually exhibited reduced affinity, but an increase in cooperativity, for the heterodimer. Importantly, these studies revealed that PAMs in the same class as VU0155041 and Lu AF21934 could weakly, but significantly, potentiate the activity of a selective mGluR2 agonist, suggesting that transactivation may occur between the two receptors within the dimer (Fig. 2c). This proposed transactivation was further supported by the ability of an mGluR2 NAM, MNI 137, to non-competitively block the response of the heterodimer to an mGluR4 agonist120.

a | Expression of metabotropic glutamate receptor type 4 (mGluR4; beige receptor) alone results in potentiation of responses to glutamate (blue circle) by PAMs (positive allosteric modulators; green hexagon) such as PHCCC and 4-PAM2 (left graph of the panel). Co-expression of mGluR2 (blue receptor) results in a loss of PHCCC- or 4-PAM2-induced potentiation of the response to glutamate (middle graph). When an mGluR2-selective agonist (red circle) is used to activate responses in cells that express mGluR2 and mGluR4, no potentiation by PHCCC or 4-PAM2 is observed (right graph). b | PAMs such as VU0155041 or Lu AF21934 (yellow hexagon), which are known to interact with a distinct site on the mGluR4 protein, are able to potentiate glutamate responses at mGluR4 homomeric receptors (left graph of the panel). In contrast to PHCCC and 4-PAM2, the potentiation of the glutamate response by VU0155041 or Lu AF21934 is retained in the presence of mGluR2 (middle graph) and these PAMs are also able to shift the response curve of an mGluR2-selective agonist markedly to the left (right graph), suggesting that there is transactivation between the two sides of the receptor dimer.
The capacity of mGluR2 and mGluR4 to form a pharmacologically unique heterodimer is especially interesting in light of previous studies that revealed that these receptors are co-expressed in specific brain circuits, such as at the synapses of neurons that originate in the cortex and project to the striatum (corticostriatal synapses). mGluR2 and mGluR4 can be co-immunoprecipitated using subtype-selective antibodies from cultured cell lines and from cortical and striatal brain tissue from mice and rats120, suggesting a potential interaction of the proteins in these regions. Interestingly, PHCCC, which potentiates mGluR4-mediated but not mGluR2–mGluR4 heterodimer-mediated responses in cell lines, did not potentiate responses to mGluR4 agonists at the corticostriatal synapse120. By contrast, both of the mGluR2–mGluR4 heterodimer PAMs — Lu AF21934 (Ref. 69) and VU0155041 (Ref. 120) — induced robust potentiation of the response to an mGluR4 agonist. The finding that no potentiation was observed with PHCCC suggested that, as in cell lines, the mGluR2–mGluR4 interaction may be dominant, particularly over mGluR4 homodimers, whenever mGluR2 and mGluR4 are co-expressed. In contrast to the lack of efficacy of PHCCC at the corticostriatal synapse, this mGluR4 PAM induces strong potentiation of responses to mGluR5 activation at several other central synapses where mGluR4 is likely to function as a homodimer85,122,123. Together with previous studies, these observations provide strong evidence that mGluR2 and mGluR4 can function as pharmacologically distinct homodimers or mGluR2–mGluR4 heterodimers, each of which has distinct functions in the CNS. This suggests that a revision of our view of mGluR function and the pharmacology of mGluR allosteric modulators is warranted, and that an increased focus on understanding mGluR heterodimerization will help to ensure that compounds with the highest potential for efficacy are developed. Additionally, it may be possible to tailor drug development to either engage or avoid heterodimers, potentially limiting adverse effects. Finally, an appreciation of the complexity of heterodimerization is needed to understand and interpret the results of studies of the effects of allosteric modulators in native systems.
Complexities in allosteric modulator optimization
The new insights into allosteric modulator function that are outlined above highlight the need to address the complexities that are associated with biased signalling, with the effects of receptor expression on ago-PAM versus pure PAM activities, with species specificity, with differential effects on affinity versus efficacy and with the potential impact of heterodimer formation. In addition, allosteric modulators have notoriously shallow SARs and exhibit fundamental changes in their modes of efficacy (known as 'molecular switches') upon minor structural changes to the scaffold19,78,124,125,126. Although these issues raise challenges, each can be managed in a practical way to allow lead-optimization efforts to advance strong candidates for development.
The propensity of structurally similar ago-PAMs, PAMs, NAMs and NALs to be produced from a given allosteric-modulator chemotype has been termed mode switching, and the subtle underlying structural changes that elicit these divergent outcomes are often referred to as molecular switches. The concept of molecular switches can also be extended to encompass small changes to a ligand that modulate its receptor-subtype selectivity26,127,128,129,130. In a lead-optimization campaign, it is important to avoid chemical series that possess a strong propensity for molecular switching, as this can complicate the SAR, and the metabolites that are generated in vivo can switch the mode of pharmacology or alter receptor-subtype selectivity29. To rapidly identify molecular switches, we and others have utilized a 'triple add' screening paradigm, for both high-throughput screening and primary screening, that allows the detection of agonist, PAM and antagonist activity in a single screen27,41. We first noted molecular switching within a series of mGluR5 NAMs that were based on a heterobiarylacetylene chemotype127,128. Here, subtle structural modifications converted a partial NAM (compound 1) into either a full NAM (compound 2) or a potent PAM (compound 3) (Fig. 3a). The fact that the addition of a small polar moiety in compound 3 had such a profound effect on pharmacology led us to evaluate the oxidative metabolites of allosteric modulators. Indeed, oxidative metabolism converted a pure mGluR5 PAM (compound 4) into a potent mGluR5 ago-PAM (compound 5) that engendered marked adverse events in vivo that were attributable solely to the switch in pharmacological mode of the metabolite to an ago-PAM29 (Fig. 3b).

a | The first report of molecular switches within the classical biarylacetylene core, where subtle changes convert an mGluR5 (metabotropic glutamate receptor type 5) partial antagonist (compound 1; which fully occupied the allosteric site but decreased an 80% maximal effector concentration, EC80, to only an EC29) to either a potent NAM (negative allosteric modulator; compound 2) or a PAM (positive allosteric modulator; compound 3). b | The first example of an in vivo cytochrome P450 (CYP)-mediated molecular switch of a pure mGluR5 PAM (compound 4) into a potent ago-PAM (compound 5) that engendered adverse events. c | Exploiting a scaffold with a propensity for molecular switches to gain access to ligands for another receptor subtype. Here, a potent mGluR5 PAM with weak mGluR3 NAM activity (compound 6) is modified, via a para-methoxy (p-OMe) molecular switch, into the first selective, potent mGluR3 NAM (compound 7) which can also penetrate the central nervous system. Glu Max, maximal response to glutamate; IC50, half-maximal inhibitory concentration.
In addition to mode switches, SARs are notoriously shallow for GPCR allosteric modulators, and subtle electronic or steric perturbation of a ligand often leads to a complete loss of activity, dramatically complicating the drug metabolism and pharmacokinetic (DMPK) and chemical optimization for clinical candidates19,78,124,125,126. The literature in this area is filled with examples in which the synthesis of thousands of compounds yields libraries in which less than 10% of the compounds are active as allosteric modulators, with subtle steric or stereoelectronic changes abolishing activity (for example, see Ref. 131); yet, there are also cases wherein the SAR is robust and tractable19,78,124,125,126. Therefore, for the chemical optimization of allosteric ligands, focused iterative library synthesis provides a distinct advantage over traditional singleton approaches; moreover, multiple dimensions of a scaffold must be surveyed to identify regions that can tolerate modification. As SARs for allosteric modulators are often extremely shallow or flat, the strategy of 'walking' fluorine atoms around an allosteric ligand has achieved some success in identifying positions that are tolerant to change; moreover, once structural changes are incorporated into the core, optimization efforts are typically more fruitful, as illustrated by the M1-receptor PAM BQCA (compound 8), and analogues 9–11 (Refs 72,132,133) (Fig. 4). It is important to consider that the composite potency value for a PAM (the effector concentration for half-maximum response; EC50) at a fixed concentration of agonist is not derived from a single ligand–receptor interaction, but rather a composite of the multiple pharmacological parameters that have been outlined above (τB, α and β) as well as the dissociation constant (pKB), all of which are subject to unique SARs19,78,124,125,126,134. Dissecting the individual contributions of each parameter to the composite EC50 of the M1-receptor PAM BQCA (compound 8) and its related analogues — a series with very flat SAR (based on composite EC50) — showed that the SAR of this series was actually robust and markedly enriched the understanding of flat allosteric SARs134. Figure 5 shows our current approach to allosteric-modulator chemistry and profiling, with a balance between driving improvements in the SAR with a primary measure of compound potency and extensively profiling select compounds to widen our understanding of compound pharmacology.

The prototypical scaffold of BQCA (compound 8), which is an M1 muscarinic acetylcholine receptor PAM (positive allosteric modulator), was shown to possess a notoriously 'flat' or 'shallow' structure–activity relationship (SAR). An initial chemical-optimization effort afforded hundreds of analogues (including compound 9) in which R1 and R2 were not fluoro substituents that had very low success rate in potentiating M1-receptor-mediated calcium mobilization in cell lines (fewer than 5% of compounds were active). Application of the 'fluorine walk' identified multiple regions in which fluorine atoms were tolerated and improved M1-receptor potency, such as in compound 10. A subsequent re-optimization campaign with the optimal fluorine atoms in place was highly productive and led to the discovery of potent M1-receptor PAMs, such as compound 11. The inset summarizes the effects of modifications of different regions of the BCQA scaffold on different components of the PAM response. EC50, effector concentration for half-maximum response; pKB, dissociation constant.

Our approach has been to drive an iterative chemistry effort primarily from the determination of the EC50 (effector concentration for half-maximum response) and maximal responses to an allosteric modulator. In the majority of cases, the most well-characterized endogenous ligand is used; in situations in which more than one endogenous ligand exists, profiling should include all relevant endogenous ligands, at least with selected compounds. Although the '% max' values that are determined in this manner may be artificially capped (that is, the maximal response induced by the compound has reached the maximal response of the system), this capping can serve as an initial triage step to rank compounds as having 'high' versus 'low' efficacy. Such ranking can be validated with an efficacy experiment in which a constant amount of compound is incubated with increasing concentrations of orthosteric ligand ('fold shift'). Selected compounds can then be progressed to produce concentration–response curves to examine the effects of multiple concentrations of allosteric modulator in the presence of a full orthosteric agonist. Application of the operational model of allosterism can then be used to predict compound affinity in the functional assay to be used for assessment. Additional profiling in other functional assays can be used to assess ligand bias; at this stage it is important to consider non-G-protein-dependent assays, such as β-arrestin recruitment. If an allosteric or orthosteric radioligand is available, compounds can be further profiled for displacement or effects on radioligand affinity. Intentional optimization of compounds with distinct in vitro profiles (signal bias, probe dependence) can then be pursued, with the goal of translating these properties into native or in vivo system models, such as for brain-slice electrophysiology or behavioural studies. Additionally, compounds with differential profiles can be tested for adverse effects to try to understand if a signalling profile that has been identified in vitro may predict efficacy and/or adverse effect profiles. Additional libraries of compounds can then be synthesized by rapidly identifying tractable structure–activity relationships (SARs) and using deuterium labelling or fluorine walk strategies to address the common finding that allosteric ligands often lose activity or switch pharmacology at the target with chemical modification. In parallel, drug metabolism and pharmacokinetics (DMPK) analyses (including the profiling of possible cytochrome-P450 (CYP) interactions and blood–brain barrier permeability) should be ongoing to identify compounds with the highest likelihood to progress.
Flat SARs represent further challenges in attempts to optimize the DMPK profile of an allosteric ligand. Whereas subtle steric or stereoelectronic modifications often abolish potency, blocking sites of oxidative metabolism or introducing chemical 'shunts' is not possible. For example, the selective mGluR3 NAM (compound 7) underwent rapid cytochrome P450 (CYP)-mediated O-dealkylation of the para-methoxy moiety, which is the essential switch for mGluR3 NAM activity64. All attempts to block or slow metabolism with steric and electronic approaches led to a loss of mGluR3 NAM activity. Ultimately, replacing the CH3 group with CD3 maintained mGluR3 activity and decreased metabolism, enabling in vivo proof-of-concept studies64. Based on the results from such studies, blocking metabolically labile sites with deuterium has emerged as a powerful tool in the allosteric medicinal-chemistry toolkit for successful optimization.
After a decade of effort in this arena, several principles have emerged for the successful chemical optimization of allosteric ligands for GPCRs19,78,124,125,126. First, it is beneficial to perform primary assays in 'triple add' mode, or to use another strategy to capture ligands with a propensity for 'molecular switches'; such ligands or chemotypes should be avoided for lead optimization. In the same vein, owing to potential species differences, the optimization screening plan needs to include cell lines from humans as well as other species, and chemotypes that display strong species bias should be deprioritized to accelerate clinical development. Second, multi-dimensional iterative parallel synthesis — as opposed to singleton synthesis — and early introduction of the fluorine walk are both highly recommended to rapidly identify productive SARs and series for advancement. Third, major metabolites of allosteric ligands must be synthesized and evaluated to ensure that no CYP-mediated molecular switches are present; if any are, the series should be discarded. Using deuterium to block metabolic 'soft' spots has emerged as an attractive strategy to improve disposition while maintaining target potency. Fourth, to avoid potential adverse events, or for instances where ago-PAM activity is desired, the chemical lead optimization should be driven in cell lines with low receptor expression and cross-checked in native systems. Fifth, in terms of stimulus bias, it is best to choose scaffolds with no evidence of stimulus bias unless the bias can be rationally understood from a biological perspective. Finally, as different allosteric ligands can act at the same, partially overlapping, or distinct allosteric sites, radio- and PET-ligand efforts should be developed in the same chemical series as the candidate and the candidate should be confirmed — using extensive in vitro profiling to ensure accurate occupancy data — to act competitively.
Conclusions
The discovery and development of allosteric modulators for GPCRs that are expressed in the CNS has advanced considerably over the past decade. The isolation of the first crystal structures of class A, class B and class C GPCR representatives, particularly in complex with allosteric ligands, will undoubtedly open new avenues for the development of therapeutics for this important family of drug targets. Careful attention to issues such as signal bias, molecular switching, the generation of pharmacologically active metabolites and complex native-tissue or in vivo pharmacology will push the field closer to the realization of allosteric modulation as a path forward for the development of therapeutics for some of the most challenging human neurological and psychiatric disorders.
What will the next decade of allosteric modulator pharmacology bring? The finding of a highly conserved binding site for allosteric compounds in the same region of many GPCRs suggests that this site — or allosteric sites in general — may not be accidental. The potential discovery of 'natural' allosteric ligands raises the question of what may be the 'true' orthosteric ligand and raises interesting queries regarding the physiological implications of the ability of synthetic small molecules or therapeutic antibodies to displace potential endogenous allosteric compounds. The observation that heterodimers with drastically different pharmacological responses exist suggests that there will be multiple opportunities to tailor drug development to specific receptor heteromers; it will be paramount to understand which combinations are required for efficacy, to ensure that these combinations are recapitulated in the human brain and to identify combinations that might be rationally avoided to limit adverse effects. Although at present the complexities of signal bias represent a challenge for the medicinal-chemistry campaigns for allosteric modulators and should be avoided until they have been better understood and translated in vivo, the field of pharmacology must take on the challenge of this fascinating area of GPCR biology to eventually translate allosteric modulators into the most highly effective and minimally toxic therapeutics. Despite the challenges and caveats in allosteric-ligand development, the advantages of allosteric modulation of GPCRs are clear and provide exciting new opportunities for the discovery of novel agents for the treatment of CNS disorders and other diseases in humans.
Note added in proof
While the present manuscript was under review, a structure of the metabotropic glutamate receptor 5 transmembrane domain in complex with the negative allosteric modulator mavoglurant was published153.
References
Chalmers, D. T. & Behan, D. P. The use of constitutively active GPCRs in drug discovery and functional genomics. Nature Rev. Drug Discov. 1, 599–608 (2002).
Lagerström, M. C. & Schiöth, H. B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nature Rev. Drug Discov. 7, 339–357 (2008).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nature Rev. Drug Discov. 5, 993–996 (2006).
Kola, I. & Landis, J. Can the pharmaceutical industry reduce attrition rates? Nature Rev. Drug Discov. 3, 711–715 (2004).
Christopoulos, A. & Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 54, 323–374 (2002).
Changeux, J. P. The concept of allosteric interaction and its consequences for the chemistry of the brain. J. Biol. Chem. 288, 26969–26986 (2013).
Möhler, H., Fritschy, J. M. & Rudolph, U. A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 300, 2–8 (2002).
Lindberg, J. S. et al. Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J. Am. Soc. Nephrol. 16, 800–807 (2005).
Harrington, P. E. & Fotsch, C. Calcium sensing receptor activators: calcimimetics. Curr. Med. Chem. 14, 3027–3034 (2007).
Dorr, P. et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49, 4721–4732 (2005).
Emmitte, K. A. mGlu5 negative allosteric modulators: a patent review (2010–2012). Exp. Opin. Ther. Pat. 23, 393–408 (2013).
Rocher, J. P. et al. mGluR5 negative allosteric modulators overview: a medicinal chemistry approach towards a series of novel therapeutic agents. Curr. Top. Med. Chem. 11, 680–695 (2011).
Lavreysen, H. et al. Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H]JNJ-40068782. J. Pharmacol. Exp. Ther. 346, 514–527 (2013).
Hopkins, C. R. Is there a path forward for mGlu2 positive allosteric modulators for the treatment of schizophrenia? ACS Chem. Neurosci. 4, 211–213 (2013).
Macdonald, G. J. & Lindsley, C. W. A unique industrial–academic collaboration towards the next generation of schizophrenia therapeutics. Curr. Top. Med. Chem. 14, 304–312 (2014).
Lindsley, C. W. & Hopkins, C. R. Metabotropic glutamate receptor 4 (mGlu4)-positive allosteric modulators for the treatment of Parkinson's disease: historical perspective and review of the patent literature. Expert Opin. Ther. Pat. 22, 461–481 (2012).
Foster, D. J., Jones, C. K. & Conn, P. J. Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov. Med. 14, 413–420 (2012).
Wootten, D., Christopoulos, A. & Sexton, P. M. Emerging paradigms in GPCR allostery: implications for drug discovery. Nature Rev. Drug Discov. 12, 630–644 (2013).
Conn, P. J., Christopoulos, A. & Lindsley, C. W. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev. Drug Discov. 8, 41–54 (2009).
O'Brien, J. A. et al. A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol. Pharmacol. 64, 731–740 (2003).
Rodriguez, A. L. et al. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol. 68, 1793–1802 (2005).
Canals, M., Sexton, P. M. & Christopoulos, A. Allostery in GPCRs: 'MWC' revisited. Trends Biochem. Sci. 36, 663–672 (2011).
Gregory, K. J., Sexton, P. M. & Christopoulos, A. Overview of receptor allosterism. Curr. Protoc. Pharmacol. Ch. 1, Unit 1.21 (2010).
Leach, K. Sexton, P. M. & Christopoulos, A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 28, 382–389 (2007).
May, L. T. et al. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51 (2007).
Gregory, K. J. et al. Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol. Pharmacol. 82, 860–875 (2012).
Niswender, C. M. et al. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 74, 1345–1358 (2008).
Noetzel, M. J. et al. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol. Pharmacol. 81, 120–133 (2012).
Rook, J. M. et al. Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol. Psychiatry 73, 501–509 (2013).
Knudsen, L. B. et al. Functional importance of GLP-1 receptor species and expression levels in cell lines. Regul. Pept. 175, 21–29 (2012).
Ayala, J. E. et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34, 2057–2071 (2009).
Spalding, T. A. et al. Discovery of an ectopic activation site on the M1 muscarinic receptor. Mol. Pharmacol. 61, 1297–1302 (2002).
Sur, C. et al. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-d-aspartate receptor activity. Proc. Natl Acad. Sci. USA 100, 13674–13679 (2003).
Jones, C. K. et al. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 28, 10422–10433 (2008).
Langmead, C. J. et al. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br. J. Pharmacol. 154, 1104–1115 (2008).
Lebois, E. P. et al. Discovery and characterization of novel subtype-selective allosteric agonists for the investigation of M1 receptor function in the central nervous system. ACS Chem. Neurosci. 1, 104–121 (2010).
Lebon, G. et al. Mutagenic mapping suggests a novel binding mode for selective agonists of M1 muscarinic acetylcholine receptors. Mol. Pharmacol. 75, 331–341 (2009).
Digby, G. J. et al. Chemical modification of the M1 agonist VU0364572 reveals molecular switches in pharmacology and a bitopic binding mode. ACS Chem. Neurosci. 3, 1025–1036 (2012).
Valant, C. et al. A novel mechanism of G protein-coupled receptor functional selectivity: muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J. Biol. Chem. 283, 29312–29321 (2008).
Kenakin, T. Allosteric modulators: the new generation of receptor antagonist. Mol. Interv. 4, 222–229 (2004).
Rodriguez, A. L. et al. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol. 78, 1105–1123 (2010).
Malherbe, P. et al. Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J. Biol. Chem. 278, 8340–8347 (2003).
Brauner-Osborne, H. Wellendorph, P. & Jensen, A. A. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr. Drug Targets 8, 169–184 (2007).
Gregory, K. J. et al. Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 60, 66–81 (2011).
Gregory, K. J. et al. Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol. Pharmacol. 83, 991–1006 (2013).
Wang, C. I. & Lewis, R. J. Emerging opportunities for allosteric modulation of G-protein coupled receptors. Biochem. Pharmacol. 85, 153–162 (2013).
Bokoch, M. P. et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463, 108–112 (2010).
Kruse, A. C. et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013).
Wu, H. et al. Structure of class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 (2014).
Hollenstein, K. et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 (2013).
Nguyen, E. D. et al. Assessment and challenges of ligand docking into comparative models of G-protein coupled receptors. PLoS ONE 8, e67302 (2013).
Dror, R. O. et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 (2013).
Avlani, V. A. et al. Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J. Biol. Chem. 282, 25677–25686 (2007).
Hoare, S. R. et al. Allosteric ligands for the corticotropin releasing factor type 1 receptor modulate conformational states involved in receptor activation. Mol. Pharmacol. 73, 1371–1380 (2008).
Niswender, C. M. & Conn, P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322 (2010).
Satoh, A. et al. Discovery and in vitro and in vivo profiles of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorg. Med. Chem. Lett. 19, 5464–5468 (2009).
Katritch, V., Cherezov, V. & Stevens, R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013).
Urwyler, S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol. Rev. 63, 59–126 (2011).
Hemstapat, K. et al. A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol. Pharmacol. 70, 616–626 (2006).
Chen, Y. et al. Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol. Pharmacol. 71, 1389–1398 (2007).
Annoura, H. et al. A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates. Bioorg. Med. Chem. Lett. 6, 763–766 (1996).
Litschig, S. et al. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol. Pharmacol. 55, 453–461 (1999).
Varney, M. A. et al. SIB-1757 and SIB-1893: selective, noncompetitive antagonists of metabotropic glutamate receptor type 5. J. Pharmacol. Exp. Ther. 290, 170–181 (1999).
Wenthur, C. J. et al. Discovery of (R)-(2-fluoro-4-((-4- methoxyphenyl)ethynyl)phenyl) (3-hydroxypiperidin- 1-yl)methanone (ML337), an mGlu3 selective and CNS penetrant negative allosteric modulator (NAM). J. Med. Chem. 56, 5208–5212 (2013).
Sheffler, D. J. et al. Allosteric modulation of metabotropic glutamate receptors. Adv. Pharmacol. 62, 37–77 (2011).
Kalinichev, M. et al. ADX71743, a potent and selective negative allosteric modulator of metabotropic glutamate receptor 7: in vitro and in vivo characterization. J. Pharmacol. Exp. Ther. 344, 624–636 (2013).
Jones, C. K. et al. The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with l-DOPA or an adenosine 2A antagonist in preclinical rodent models of Parkinson's disease. J. Pharmacol. Exp. Ther. 340, 404–421 (2012).
Le Poul, E. et al. A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson's disease. J. Pharmacol. Exp. Ther. 343, 167–177 (2012).
Bennouar, K. E. et al. Synergy between l-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson's disease treatment and dyskinesia. Neuropharmacology 66, 158–169 (2013).
Brady, A. E. et al. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 327, 941–953 (2008).
Byun, N. E. et al. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology 39, 1578–1593 (2014).
Ma, L. et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl Acad. Sci. USA 106, 15950–15955 (2009).
Marlo, J. E. et al. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol. 75, 577–588 (2009).
Shirey, J. K. et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci. 29, 14271–14286 (2009).
Chan, W. Y. et al. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc. Natl Acad. Sci. USA 105, 10978–10983 (2008).
Shirey, J. K. et al. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nature Chem. Biol. 4, 42–50 (2008).
Bridges, T. M. et al. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J. Med. Chem. 52, 3445–3448 (2009).
Melancon, B. J. et al. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J. Med. Chem. 55, 1445–1464 (2012).
Digby, G. J. et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J. Neurosci. 32, 8532–8544 (2012).
Valant, C. et al. Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol. Pharmacol. 81, 41–52 (2012).
Suratman, S. et al. Impact of species variability and 'probe-dependence' on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br. J. Pharmacol. 162, 1659–1670 (2011).
Cho, H. P. et al. A novel class of succinimide-derived negative allosteric modulators of metabotropic glutamate receptor subtype 1 provides insight into a disconnect in activity between the rat and human receptors. ACS Chem. Neurosci. http://dx.doi.org/10.1021/cn5000343 (2014).
Parmentier-Batteur, S. et al. Mechanism based neurotoxicity of mGlu5 positive allosteric modulators — development challenges for a promising novel antipsychotic target. Neuropharmacology 82, 161–173 (2013).
Nickols, H. H. & Conn, P. J. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis. 61, 55–71 (2014).
Marino, M. J. et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc. Natl Acad. Sci. USA 100, 13668–13673 (2003).
Kenakin, T. & Christopoulos, A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nature Rev. Drug Discov. 12, 205–216 (2013).
Digby, G. J., Conn, P. J. & Lindsley, C. W. Orthosteric- and allosteric-induced ligand-directed trafficking at GPCRs. Curr. Opin. Drug Discov. Devel. 13, 587–594 (2010).
López De Jesús, M. et al. Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J. Biol. Chem. 281, 21837–21847 (2006).
Leach, K. et al. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 35, 855–869 (2010).
Zhang, Y., Rodriguez, A. L. & Conn, P. J. Allosteric potentiators of metabotropic glutamate receptor subtype 5 have differential effects on different signaling pathways in cortical astrocytes. J. Pharmacol. Exp. Ther. 315, 1212–1219 (2005).
Sheffler, D. J. & Conn, P. J. Allosteric potentiators of metabotropic glutamate receptor subtype 1a differentially modulate independent signaling pathways in baby hamster kidney cells. Neuropharmacology 55, 419–427 (2008).
Leach, K. et al. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology 153, 4304–4316 (2012).
Davey, A. E. et al. Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153, 1232–1241 (2012).
Ahn, K. H., Mahmoud, M. M. & Kendall, D. A. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J. Biol. Chem. 287, 12070–12082 (2012).
Niswender, C. M. et al. Context-dependent pharmacology exhibited by negative allosteric modulators of metabotropic glutamate receptor 7. Mol. Pharmacol. 77, 459–468 (2010).
Mathiesen, J. M. et al. Identification of indole derivatives exclusively interfering with a G protein-independent signaling pathway of the prostaglandin D2 receptor CRTH2. Mol. Pharmacol. 68, 393–402 (2005).
Maillet, E. L. et al. A novel, conformation-specific allosteric inhibitor of the tachykinin NK2 receptor (NK2R) with functionally selective properties. FASEB J. 21, 2124–2134 (2007).
Noetzel, M. J. et al. A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Mol. Pharmacol. 83, 835–847 (2013).
Goshal, A. et al. Stimulus bias of metabotropic glutamate receptor 5 allosteric modulators — impact on CNS effects and implications for therapeutic use. Program No. 614.01. 2013 Neuroscience Meeting Planner. (San Diego, CA; Society for Neuroscience, 2013).
Price, M. R. et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol. 68, 1484–1495 (2005).
Wang, X. et al. Effects of the allosteric antagonist 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea (PSNCBAM-1) on CB1 receptor modulation in the cerebellum. Mol. Pharmacol. 79, 758–767 (2011).
Ayoub, M. A. et al. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 21522–21528 (2002).
Ayoub, M. A. et al. Preferential formation of MT1/MT2 melatonin receptor heterodimers with distinct ligand interaction properties compared with MT2 homodimers. Mol. Pharmacol. 66, 312–321 (2004).
Ferre, S. et al. Adenosine A1 receptor-mediated modulation of dopamine D1 receptors in stably cotransfected fibroblast cells. J. Biol. Chem. 273, 4718–4724 (1998).
Milligan, G. The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol. Pharmacol. 84, 158–169 (2013).
Rozenfeld, R. & Devi, L. A. Exploring a role for heteromerization in GPCR signalling specificity. Biochem. J. 433, 11–18 (2011).
Ferre, S. et al. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol. Rev. 66, 413–434 (2014).
Kostenis, E. et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 111, 1806–1813 (2005).
AbdAlla, S. et al. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 276, 39721–39726 (2001).
Levoye, A. et al. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J. 25, 3012–3023 (2006).
Milligan, G. The role of dimerisation in the cellular trafficking of G-protein-coupled receptors. Curr. Opin. Pharmacol. 10, 23–29 (2010).
Archbold, J. K. et al. Structural insights into RAMP modification of secretin family G protein-coupled receptors: implications for drug development. Trends Pharmacol. Sci. 32, 591–600 (2011).
Pin, J. P. & Prezeau, L. Allosteric modulators of GABAB receptors: mechanism of action and therapeutic perspective. Curr. Neuropharmacol. 5, 195–201 (2007).
Kniazeff, J. et al. Dimers and beyond: the functional puzzles of class C GPCRs. Pharmacol. Ther. 130, 9–25 (2011).
Romano, C., Yang, W. L. & O'Malley, K. L. Metabotropic glutamate receptor 5 is a disulfide-linked dimer. J. Biol. Chem. 271, 28612–28616 (1996).
Doumazane, E. et al. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 25, 66–77 (2011).
Kammermeier, P. J. Functional and pharmacological characteristics of metabotropic glutamate receptors 2/4 heterodimers. Mol. Pharmacol. 82, 438–447 (2012).
Lundstrom, L. et al. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br. J. Pharmacol. 164, 521–537 (2011).
Hlavackova, V. et al. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J. 24, 499–509 (2005).
Yin, S. et al. Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci. 34, 79–94 (2014).
Ortuno, D. et al. Identification and characterization of a potent and selective positive allosteric modulator of mGluR4. Program No. 823.27. 2008 Neuroscience Meeting Planner. (Washington, DC; Society for Neuroscience, 2008).
Jones, P. J., Xiang, Z. & Conn, P. J. Metabotropic glutamate receptors mGluR4 and mGluR8 regulate transmission in the lateral olfactory tract-piriform cortex synapse. Neuropharmacology 55, 440–446 (2008).
Valenti, O. et al. Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J. Pharmacol. Exp. Ther. 313, 1296–1304 (2005).
Melancon, B. J. et al. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer's disease. Drug Discov. Today 18, 1185–1199 (2013).
Menniti, F. S. et al. Allosteric modulators for the treatment of schizophrenia: targeting glutamatergic networks. Curr. Top. Med. Chem. 13, 26–54 (2013).
Wenthur, C. J. et al. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 54, 165–184 (2014).
Wood, M. R. et al. “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry 50, 2403–2410 (2011).
Sharma, S. et al. Synthesis and SAR of an mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg. Med. Chem. Lett. 18, 4098–4101 (2008).
Sharma, S. et al. Discovery of molecular switches that modulate modes of metabotropic glutamate receptor subtype 5 (mGlu5) pharmacology in vitro and in vivo within a series of functionalized, regioisomeric 2- and 5-(phenylethynyl)pyrimidines. J. Med. Chem. 52, 4103–4106 (2009).
Schann, S. et al. Chemical switch of a metabotropic glutamate receptor 2 silent allosteric modulator into dual metabotropic glutamate receptor 2/3 negative/positive allosteric modulators. J. Med. Chem. 53, 8775–8779 (2010).
Zhao, Z. et al. Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg. Med. Chem. Lett. 17, 1386–1391 (2007).
Shirey, J. et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci. 29, 14271–14286 (2009).
Yang, F. V. et al. Parallel synthesis of N-biaryl quinolone carboxylic acids as selective M1 positive allosteric modulators. Bioorg. Med. Chem. Lett. 20, 531–536 (2010).
Mistry, S. N. et al. Synthesis and pharmacological profiling of analogues of benzyl quinolone carboxylic acid (BQCA) as allosteric modulators of the M1 muscarinic receptor. J. Med. Chem. 56, 5151–5172 (2013).
Gjoni, T. & Urwyler, S. Receptor activation involving positive allosteric modulation, unlike full agonism, does not result in GABAB receptor desensitization. Neuropharmacology 55, 1293–1299 (2008).
Sanger, H. et al. Pharmacological profiling of native group II metabotropic glutamate receptors in primary cortical neuronal cultures using a FLIPR. Neuropharmacology 66, 264–273 (2013).
Parmentier-Batteur, S. et al. Differential effects of the mGluR5 positive allosteric modulator CDPPB in the cortex and striatum following repeated administration. Neuropharmacology 62, 1453–1460 (2012).
Li, X. et al. Repeated dosing with oral allosteric modulator of adenosine A1 receptor produces tolerance in rats with neuropathic pain. Anesthesiology 100, 956–961 (2004).
Mukherjee, S. & Manahan-Vaughan, D. Role of metabotropic glutamate receptors in persistent forms of hippocampal plasticity and learning. Neuropharmacology 66, 65–81 (2013).
Gasparini, F. et al. 2-methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503 (1999).
Huber, K. M., Roder, J. C. & Bear, M. F. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J. Neurophysiol. 86, 321–325 (2001).
Bear, M. F., Huber, K. M. & Warren, S. T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377 (2004).
Lecourtier, L. et al. Positive allosteric modulation of metabotropic glutamate 5 (mGlu5) receptors reverses N-methyl-d-aspartate antagonist-induced alteration of neuronal firing in prefrontal cortex. Biol. Psychiatry 62, 739–746 (2007).
Darrah, J. M., Stefani, M. R. & Moghaddam, B. Interaction of N-methyl-d-aspartate and group 5 metabotropic glutamate receptors on behavioral flexibility using a novel operant set-shift paradigm. Behav. Pharmacol. 19, 225–234 (2008).
Liu, F. et al. ADX47273 [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J. Pharmacol. Exp. Ther. 327, 827–839 (2008).
Balschun, D., Zuschratter, W. & Wetzel, W. Allosteric enhancement of metabotropic glutamate receptor 5 function promotes spatial memory. Neuroscience 142, 691–702 (2006).
Reichel, C. M. et al. Loss of object recognition memory produced by extended access to methamphetamine self-administration is reversed by positive allosteric modulation of metabotropic glutamate receptor 5. Neuropsychopharmacology 36, 782–792 (2011).
Xu, J. et al. Potentiating mGluR5 function with a positive allosteric modulator enhances adaptive learning. Learn. Mem. 20, 438–445 (2013).
Gregory, K. J. et al. N-aryl piperazine metabotropic glutamate receptor 5 positive allosteric modulators possess efficacy in preclinical models of NMDA hypofunction and cognitive enhancement. J. Pharmacol. Exp. Ther. 347, 438–457 (2013).
Gastambide, F. et al. Selective remediation of reversal learning deficits in the neurodevelopmental MAM model of schizophrenia by a novel mGlu5 positive allosteric modulator. Neuropsychopharmacology 37, 1057–1066 (2012).
Gastambide, F. et al. The mGlu5 positive allosteric modulator LSN2463359 differentially modulates motor, instrumental and cognitive effects of NMDA receptor antagonists in the rat. Neuropharmacology 64, 240–247 (2013).
Wong, R. K. et al. Group I mGluR-induced epileptogenesis: distinct and overlapping roles of mGluR1 and mGluR5 and implications for antiepileptic drug design. Epilepsy Curr. 5, 63–68 (2005).
Doré, A. S. et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 (2014).
Acknowledgements
The authors acknowledge the following grant support: US National Institutes of Health (NIH) grants U01 MH087956, R01 MH062646, R01 MH073676, U19 MH097056, R01 NS031373, P50 NS071669 (to P.J.C.); R01 DA023947, R01 MH082867, U54 MH084659, U19 MH097056 (to C.W.L.); NIH grants R01 GM080403, R01 MH090192, R01 GM099842, R01 DK097376) and NSF grants BIO Career 0742762 and CHE 1305874 (to J.M.); NIH grant R21 NS078262, a Basic Research Grant from the International Rett Syndrome Foundation, and a Treatment Award from Autism Speaks (to C.M.N). Vanderbilt Specialized Chemistry Center is in the Molecular Libraries Probe Centers Network.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
P.J.C., C.W.L. and C.M.N. receive research support from Bristol-Myers Squibb and AstraZeneca. Each of these three authors is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic receptor allosteric modulators and have been licensed by Vanderbilt University to AstraZeneca, Bristol-Myers Squibb, and Johnson & Johnson. C.W.L. has consulted for AbbVie and received compensation. Over the past 2 years, P.J.C. has consulted for and received compensation from Pfizer, Cambridge, Massachusetts, USA.
Related links
Further information
Supplementary information
Supplementary information S1 (table)
Reported allosteric modulators of family A G protein-coupled receptors (PDF 204 kb)
Supplementary information S2 (table)
Reported allosteric modulators of family B G-protein-coupled receptors (PDF 148 kb)
Supplementary information S3 (table)
Reported allosteric modulators of family C G-protein-coupled receptors (PDF 228 kb)
Glossary
- Cooperativity factors
-
Parameters that are used to quantify the magnitude of the effect of an allosteric ligand on the affinity or functional response to the orthosteric ligand.
Rights and permissions
About this article

Cite this article
Conn, P., Lindsley, C., Meiler, J. et al. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat Rev Drug Discov 13, 692–708 (2014). https://doi.org/10.1038/nrd4308
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd4308
This article is cited by
-
Preventing incubation of drug craving to treat drug relapse: from bench to bedside
Molecular Psychiatry (2023)
-
The pyridazine heterocycle in molecular recognition and drug discovery
Medicinal Chemistry Research (2023)
-
Recent advances in dopaminergic strategies for the treatment of Parkinson’s disease
Acta Pharmacologica Sinica (2020)
-
Lrrc7 mutant mice model developmental emotional dysregulation that can be alleviated by mGluR5 allosteric modulation
Translational Psychiatry (2019)
-
GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures
Nature Reviews Drug Discovery (2019)
