
Abstract
The mood stabilizers lithium, valproate and lamotrigine are traditionally used to treat bipolar disorder. However, accumulating evidence suggests that these drugs have broad neuroprotective properties and may therefore be promising therapeutic agents for the treatment of neurodegenerative diseases, including stroke. Lithium, valproate and lamotrigine exert protective effects in diverse experimental stroke models by acting on their respective primary targets, ie, glycogen synthase kinase-3, histone deacetylases and voltage-gated sodium channels, respectively. This article reviews the most recent findings regarding the underlying mechanisms of these phenomena, which will pave the way for clinical investigations that use mood stabilizers to treat stroke. We also propose several future research avenues that may extend our understanding of the benefits of lithium, valproate and lamotrigine in improving stroke outcomes.
Similar content being viewed by others


Introduction
Stroke is a major cause of serious long-term disability in adults and the second leading cause of death worldwide. Approximately 85% of all strokes are ischemic1. To date, thrombolysis with intravenous recombinant tissue plasminogen activator (rtPA) is the only Food and Drug Administration (FDA)-approved treatment for acute ischemic stroke in the United States2. Thrombolysis improves patient outcome by facilitating reperfusion of ischemic brain tissue before irreversible damage occurs and must be administered within 4.5 h of symptom onset3. Given this narrow therapeutic window, as well as the associated risk of intracerebral hemorrhage4, the estimated rate of rtPA use is quite low, only 1.8% to 2.1% of all ischemic stroke patients5. Thus, there is an urgent need to develop novel treatment options for stroke.
A promising therapeutic direction has emerged from an unexpected field: psychiatry. The mood stabilizing drugs lithium, valproate (VPA) and lamotrigine (see Figure 1 for chemical structures) are FDA-approved treatments for bipolar disorder (BD). This severe psychiatric illness affects 1%-3% of the world's population and is characterized by cyclic episodes of mania and depression. While the etiology of BD remains elusive, increasing evidence suggests that it is associated with neuroanatomical and neurophysiological anomalies, neurocognitive deficits, and dysregulated signaling pathways and gene expression6. Lithium, which has a well-established safety profile, has been used to treat BD for more than 60 years. It is an effective treatment for mania and is also used as prophylactic therapy to prevent the recurrent manic and depressive episodes that characterize BD. Anticonvulsants, notably VPA and lamotrigine, are more effective than lithium in treating rapid-cycling bipolar patients. While lamotrigine has more robust effects against bipolar depression, VPA, like lithium, has strong anti-manic effects but is less effective in treating depressive episodes. Although these mood stabilizers have long been the mainstay drugs for BD, their underlying therapeutic mechanisms remain largely unknown.

The chemical structures of lithium, VPA and lamotrigine.
Brain imaging and postmortem investigations have demonstrated regional brain volume reductions, decreased cell number and density, and neuronal atrophy in patients with BD7, 8. Structural imaging studies have also shown that patients with BD have a particular pattern of brain abnormalities that includes widespread cortical thinning; morphological abnormalities in the amygdala, basal ganglia and thalamus; reduced gray matter volume in the prefrontal cortex, ventral striatum, and hippocampus; and enlarged lateral ventricles9, 10. Notably, chronic treatment with lithium was found to significantly increase brain gray matter volume in BD patients11. Another study found that VPA-treated patients had larger cingulate gyrus volumes, including both cortex and white matter, than unmedicated BD patients12. In light of these observations, the beneficial effects of mood stabilizing drugs are now recognized to involve neurotrophic and neuroprotective mechanisms with broad applications beyond the treatment of psychiatric illnesses. The extant evidence suggests that these mood stabilizing drugs activate neurotrophic and neuroprotective pathways, highlighting their therapeutic potential for the treatment of a variety of neurodegenerative diseases, including stroke13, 14, 15.
In this article, we review the recent findings gleaned from experimental stroke models, with a focus on ischemic stroke, regarding the protective effects of lithium, VPA and lamotrigine. To date, there is a larger body of evidence from these models supporting the protective effects of lithium and VPA compared to lamotrigine. We will address the mechanisms and effects of each mood stabilizer as they are currently understood, and point to areas where further research may extend the potential therapeutic use of mood stabilizing drugs in stroke treatment.
The pathophysiology of ischemic stroke
Ischemic stroke is caused by reduced blood supply to an area of the brain, which initiates an ischemic cascade. Ischemic stroke accounts for 85% of all stroke cases1. The pathophysiology of ischemic stroke is complex and involves both early and late phase processes. In the acute phase after stroke, the reduced blood supply depletes oxygen and glucose within the brain and prevents adenosine triphosphate (ATP) production. In turn, ATP depletion impairs ATP-dependent ion pumps and alters Na+ concentration gradients across neuronal membranes. As a result, glutamate transporters fail, causing excessive synaptic release and accumulation of glutamate. Through interaction with N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxanole propionate (AMPA) receptors, glutamate overstimulation leads to Ca2+ influx, which then drives the activation of damaging Ca2+-mediated intracellular enzymes such as calpains and caspases. These ultimately result in mitochondrial failure, generation of reactive oxygen species, and cell necrosis and apoptosis16, 17. Furthermore, the osmotic gradient caused by the ion imbalance leads to an influx of water, which triggers cytotoxic and vasogenic edema18.
Neuroinflammation has been increasingly implicated in injury progression following stroke. Post-ischemic inflammation is a dynamic process involving a complicated set of interactions between inflammatory cells and molecules19. Within hours after ischemic onset, circulating leukocytes adhere to vessel walls, infiltrate ischemic brain tissue, and amplify inflammatory signal cascades, which enhance tissue damage20, 21, 22. Microglia, the resident macrophages of the brain, are activated after stroke23, 24. Activated microglia release additional inflammatory mediators that increase the vulnerability of neurons, stimulate the expression of adhesion molecules, and mediate the adherence and infiltration of leukocytes20. Astrocytes are also activated and express cytokines, chemokines and inducible nitric oxide synthase (iNOS)25. These cytotoxic factors induce further cell damage, as well as disrupt the blood-brain barrier (BBB) to further exacerbate brain injury and cause brain edema26.
The BBB is composed of capillary endothelial cells and pericytes, which are surrounded by basal lamina, astrocytic end-feet, and perivascular interneurons27. These endothelial cells are stitched together by tight junctions, which act as a highly selective and effective physical barrier to protect the CNS from pathogens and harmful molecules in the bloodstream. Tight junctions consist of transmembrane proteins, including claudins, occludins, and adhesion junction molecules, as well as cytoplasmic accessory proteins such as zonula occludens-1 (ZO-1)27. Impairment of tight junctions disrupts BBB integrity and increases its permeability. BBB disruption is a critical event in the pathogenesis of various neurological disorders, including stroke. Loss of BBB integrity following stroke allows the penetration of intravascular proteins and fluid into the cerebral parenchymal extracellular space, resulting in leukocyte infiltration, vasogenic edema and hemorrhage. Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that have multiphasic roles in ischemic stroke28. Within hours to days after cerebral ischemia, high levels of MMPs, especially MMP-2 and 9, degrade tight junctions and basal lamina proteins, disrupt cell-matrix homeostasis, and breach BBB integrity28, 29. In rodent models of ischemic stroke, both MMP-2 and 9 are linked with increased BBB permeability in the acute phase after ischemic stroke (within hours)30, 31, whereas MMP-9 is responsible for the severe BBB disruption that occurs at 24 to 48 h after ischemia32. Similarly, BBB disruption at approximately 24 h after acute stroke onset in humans is associated with increased plasma MMP-933. Elevated leukocytic MMP-9 is also closely tied to BBB disruption, as it promotes the degradation of the basal lamina in human brain tissue after stroke34. The functional significance of this enzyme has been demonstrated in mice, where genetic knockout of MMP-9 or suppression with an MMP inhibitor confers resistance to cerebral ischemia-induced BBB disruption by preventing degradation of the tight junction protein ZO-135, 36.
Neurovascular repair and regeneration take place in the chronic phase after stroke and determine the ultimate extent of the damage. The primary neurovascular responses during stroke recovery involve angiogenesis and neurogenesis. Angiogenesis is the process by which new capillaries are formed through directed proliferation and migration of endothelial progenitor cells from pre-existing blood vessels. Neurogenesis is the process of generating new neurons from neural stem and progenitor cells. Increasing evidence gleaned from human stroke patients and experimental stroke models suggests that endogenous angiogenesis and neurogenesis occur in the penumbra region days to weeks after stroke onset37. A close link between these two processes exists following stroke, as they share growth factors and signaling pathways for cell formation and migration, as well as common physical space as a result of parallel anatomic patterning and development38. Pharmacological therapies that enhance endogenous angiogenesis and neurogenesis following stroke have been shown to promote the recovery of neurological function in experimental stroke models39, 40, 41. Thus, enhancement of angiogenesis and neurogenesis may be a promising avenue for improving post-stroke clinical outcomes and functional recovery.
Genes participating in post-ischemic neurovascular regeneration have been extensively studied in experimental stroke models. Vascular endothelial growth factor (VEGF) and MMPs are two key factors involved in this neurovascular recovery process, and both have biphasic functions in the neurovascular response to stroke. VEGF and MMPs increase BBB permeability in the acute phase but accelerate angiogenesis and neurogenesis in the later phases42. In the delayed phase of ischemic recovery, VEGF can induce proliferation, inhibit apoptosis, and increase survival in both endothelial and neural cells43, 44, while MMPs can interact with extracellular matrix and growth factors or cytokine substrates to modulate neurovascular plasticity28. In addition, a feedback loop may exist between VEGF and MMPs, in which MMPs process pro-forms of matrix-bound VEGF into freely diffusible bioactive forms of VEGF45. This notion is based on findings from a mouse stroke model showing that MMP inhibition reduced endogenous VEGF signals, whereas exogenous VEGF prevented MMP inhibition-induced infarct exacerbation46.
As we review below, accumulating evidence demonstrates that the mood stabilizers lithium, VPA and lamotrigine exert protective effects throughout the pathophysiological processes of stroke.
The effects of mood stabilizing drugs in experimental stroke models
Lithium
Therapeutic benefits
The neuroprotective effects of lithium against cerebral ischemia were first demonstrated by Nonaka and Chuang in a rat model of focal cerebral ischemia47. Chronic pretreatment with lithium significantly improved neurological deficit scores and reduced brain infarct volume in ischemic animals. In a gerbil global cerebral ischemic model, lithium pretreatment at 3 mEq/kg for seven days suppressed neuronal cell death in the hippocampal CA1 region and protected against hyperactivity and memory impairment48. In addition to pre-insult treatment, post-insult lithium administration at therapeutically relevant doses (ie, 0.5 or 1.0 mEq/kg, sc) has also been shown to reduce brain infarct volume and facilitate neurological (motor, sensory and reflex) recovery in a rat model of middle cerebral artery occlusion (MCAO)49. It is worth noting that because lithium offers beneficial effects when administered at least three hours after ischemic onset, it has great clinical potential for the treatment of acute stroke49.
Primary target: glycogen synthase kinase-3
Pioneering studies demonstrated that lithium directly inhibits glycogen synthase kinase-3 (GSK-3)50, 51. This enzyme has been further established as a crucial target for lithium's cellular effects13, 52. GSK-3, consisting of α and β isoforms, is a serine/threonine kinase that regulates diverse cellular and neurophysiological processes. Lithium competes with magnesium to directly inhibit GSK-3 by binding to the active site of the enzyme and limiting its catalytic activity53. Lithium also indirectly inhibits GSK-3 activity by enhancing phosphorylation of GSK-3α at Ser21 and GSK-3β at Ser9 via activation of phosphatidylinositol 3-kinase (PI3-K)/Akt, protein kinase A and protein kinase C13, 54. In addition, lithium has been shown to increase the activities of two transcription factors, activator protein-1 (AP-1) and cyclic AMP-response element binding protein (CREB), both in vivo and in vitro55. Lithium also activates the mitogen-activated protein (MAP) kinase pathway56.
Anti-excitotoxic effects
The beneficial effects of lithium in experimental cerebral ischemia may partially occur through the inhibition of excessive NMDA receptor-mediated Ca2+ influx into neurons57. Specifically, brain ischemia stimulates NMDA receptor subunit 2A (NR2A) and NR2B activation by increasing tyrosine phosphorylation58. Lithium has been shown to reduce the activation of NR2A and 2B by inhibiting tyrosine phosphorylation without affecting total protein levels59, 60. Furthermore, because long-term lithium treatment was required for robust neuroprotection, these findings suggest that the anti-excitotoxic effects of lithium also involve altered signaling pathways and gene expression. Supporting this notion, long-term pretreatment with lithium was found to enhance brain-derived neurotrophic factor (BDNF)/TrkB signaling, activate the PI3-K/Akt cascade, and suppress the activation of MAP kinases in cultured neurons under glutamate challenge56, 61, 62. Akt has been shown to induce phosphorylation and inactivation of BAD63, a pro-apoptotic member of the B-cell lymphoma 2 (Bcl-2) family, and enhance the expression of Bcl-264, an antiapoptotic protein. Consistent with its protective effects, lithium treatment promoted Bcl-2 expression but reduced the expression of p53 and Bax in rat cerebellar granule cells under glutamate challenge, thus inhibiting caspase activation65. Lithium-mediated Akt activation after glutamate exposure also indirectly inhibited GSK-3 by increasing its serine phosphorylation level61. Therefore, modulation of Akt activity appears to play a key role in the neuroprotective effects of lithium against glutamate excitotoxicity.
Anti-apoptotic effects
Following stroke, Ca2+ influx leads to the release of pro-apoptotic proteins from the mitochondria to the cytoplasm, thereby inducing apoptosis66. Two enzymes activated after diverse apoptotic insults are c-Jun N-terminal kinase (JNK) and p38 MAP kinase67. These molecules can act synergistically to enhance the binding activity of AP-1, a dimeric transcription factor involved in the cellular response to stress factors and other signals68. Chronic lithium pretreatment attenuated glutamate-induced apoptosis in rat cerebellar granule cells in part by inhibiting the activation of JNK and p38, as well as the subsequent increase in AP-1 binding activity56. In a rat focal cerebral ischemia model, chronic pretreatment with lithium suppressed apoptosis in the ischemic penumbra by reducing caspase-3 immunoreactivity, DNA fragmentation, and expression of the AP-1 family member c-Jun69. These effects may be linked to lithium's ability to reverse glutamate-induced downregulation of the anti-apoptotic protein Bcl-2 and overexpression of the pro-apoptotic proteins p53 and Bax65.
Calpain is a nonlysosomal Ca2+-dependent intracellular cysteine protease, and Ca2+ influx-induced calpain over-activation has been implicated in ischemic brain injury70. A recent study found that post-insult lithium treatment mitigated apoptosis and brain damage by preventing GSK-3β and ERK dephosphorylation, reducing the activation of calpain and caspase-3, and inhibiting mitochondrial release of apoptosis-inducing factor and cytochrome c in a neonatal hypoxic-ischemic rat model71. Heat-shock protein 70 (HSP70) is a well-known cytoprotective factor that inhibits apoptosis and is induced in the ischemic penumbra where neuronal recovery takes place. Post-insult treatment with lithium increased the DNA binding activity of heat-shock factor-1 (HSF-1) to the heat-shock element, superinducing HSP70 in neurons to inhibit brain ischemia-induced apoptosis49. Lithium's ability to inhibit GSK-3 is likely associated with HSF-1 activation and HSP70 induction72. These findings suggest that lithium-induced GSK-3 inhibition contributes to the anti-apoptotic effects of the drug.
Effects on autophagy
Autophagy is a key intracellular catabolic process that involves the bulk degradation of damaged cytoplasmic proteins or organelles73. Recently, autophagic activity was shown to be increased at lesion sites after cerebral ischemia74. Lithium decreased inositol 1,4,5-trisphosphate levels by inhibiting phosphoinositol phosphatases, a process recently identified as a novel mechanism for inducing autophagy75. In contrast, lithium was also shown to reduce autophagy during the late recovery stage after neonatal hypoxia-ischemia71. Autophagy may therefore have either deleterious or beneficial effects, depending on the specific cellular context and the stage of the pathological process76. Therefore, the role of autophagy in the neuroprotective effects of lithium remains to be elucidated.
Anti-inflammatory effects
The anti-inflammatory effects of lithium in stroke are not well-established. One recent study documented the anti-inflammatory effects of lithium in a neonatal rat hypoxic-ischemic model. Post-insult lithium treatment significantly reduced total tissue loss following hypoxia-ischemia. This beneficial effect of lithium was associated with a suppression of microglial activation and an attenuation in the levels of pro-inflammatory cytokines and chemokines, eg, IL-1β and chemokine ligand 277. Using a model of lipopolysaccharide (LPS)-induced inflammation in cultured astrocytes, one study found that IL-6 production and its potentiation by interferon gamma (IFNγ) were significantly suppressed by treatment with lithium, GSK-3β siRNA, and four other GSK-3 inhibitors 78. In addition, lithium pretreatment attenuated astrocyte activation and the increase of IL-6 levels in the cerebral cortex and cerebellum of mice treated with LPS78. These findings suggest that lithium may exert anti-inflammatory effects via GSK-3 inhibition, a possibility that warrants further study in the context of stroke.
Pro-angiogenic effects
Several lines of evidence support the pro-angiogenic effects of lithium in stroke models. A functional MRI (fMRI) study in rats demonstrated that delayed administration (12 h after ischemic onset) of a therapeutic dose of lithium (1 mEq/kg, sc) followed by daily injections for two weeks enhanced the mean blood oxygenation level dependence (BOLD) response and task-related functional change of cerebral blood volume (fCBV) in the ipsilateral somatosensory cortex79. This chronic lithium treatment also significantly increased the distribution and size of the microvasculature as well as the number of GFAP-positive astrocytes, and upregulated MMP-9 expression in both endothelial cells and astrocytes in the penumbra brain regions79. These findings suggest that lithium-induced vascular transformation may contribute to the improved outcomes noted with fMRI. In a follow-up in vitro study, lithium was shown to increase Ser9 phosphorylation of GSK-3β in a concentration-dependent manner and to promote the expression and secretion of VEGF, but not BDNF, in both endothelial cells and astrocytes80. In endothelial cells, VEGF upregulation by lithium was mimicked by SB216763, another GSK-3β inhibitor, whereas PI3-K inhibition by LY294002 suppressed the lithium-induced increase in GSK-3β phosphorylation and VEGF protein levels80. However, VEGF levels in astrocytes were not affected by inhibition of either GSK-3β or PI3-K80. These data suggest that lithium promotes VEGF expression through distinct pathways in brain endothelium and astrocytes. Although these initial in vitro findings require further confirmation, they suggest that growth factor signaling may contribute to lithium's ability to promote neurovascular remodeling during stroke recovery.
Pro-neurogenic effects
As noted above, evidence suggests that neurogenesis contributes to the therapeutic actions of mood stabilizers. In hippocampal neural progenitor/precursor cell cultures, lithium increased proliferation, decreased apoptosis, and selectively enhanced the neuronal differentiation of these cells81, 82. Lithium treatment enhanced ERK and CREB phosphorylation in cultured hippocampal neural progenitor cells; PD98059, a MEK inhibitor, significantly decreased lithium-induced neuronal subtype differentiation82. In animal studies, chronic lithium administration enhanced neurogenesis in the dentate gyrus of adult mice by increasing cell proliferation and neuronal differentiation83. In a neonatal rat hypoxic-ischemic model, lithium treatment increased both proliferation and survival of neural stem and progenitor cells77. The pro-neurogenic effects of lithium have also been demonstrated in a transient global ischemic model, where they contribute to long-term beneficial effects. In this transient four-vessel occlusion model, lithium treatment increased the generation and survival of newborn cells in the hippocampal dentate gyrus and improved spatial learning and memory deficits of ischemic rats84. ERK1/2 phosphorylation following ischemia was enhanced by lithium treatment, whereas the ERK1/2 inhibitor U0126 abolished the effects of lithium on increased newborn cell generation and survival as well as behavioral improvement84. Research suggests that brain ischemia-induced neurogenesis involves the activation of receptor tyrosine kinases by inducing growth factors (eg, nerve growth factor, BDNF and VEGF) and subsequent stimulation of the downstream PI3-K/AKT and ERK pathways85. Taken together, these in vitro and in vivo findings suggest that lithium promotes post-ischemic hippocampal neurogenesis through ERK1/2-dependent cell signaling pathways and that this process plays an important role in the behavioral improvement seen following lithium treatment in ischemia.
Alternate findings
It is worth noting that a few early studies failed to observe a therapeutic benefit of lithium treatment in cerebral ischemia. For example, one report showed that lithium pretreatment at 5 mEq/kg up to two days prior to ischemia did not prevent the decrease in neuronal density in the hippocampal CA1 region of gerbils subjected to global cerebral ischemia86. The discrepancy could be related to the specific lithium treatment conditions and highlights the importance of optimizing dose and timing to maximize the protective effects of lithium and other drugs in stroke therapy.
VPA
Therapeutic benefits
Pioneering work from our laboratory demonstrated that post-insult treatment with VPA (300 mg/kg) reduced brain infarct size and improved functional outcome in both transient and permanent focal cerebral ischemic rat models87, 88, with a beneficial time window of at least three hours after ischemic onset88. Research from another lab confirmed that pretreatment with VPA prior to ischemia attenuated ischemic brain damage and the neurologic deficit in both transient and permanent focal cerebral ischemic mouse models, whereas treatment with VPA immediately after reperfusion reduced the infarct area only in the transient model89. Multiple mechanisms are involved in the protective action of VPA against cerebral ischemic injury.
Primary target: histone deacetylases
Studies conducted a decade ago identified histone deacetylases (HDACs) as direct targets of VPA, which is a fatty acid derivative90, 91. In humans, HDACs can be divided into four major classes14. At clinically relevant levels, VPA binds directly to HDAC active sites to inhibit class I (HDAC1, 2, 3, and 8 isoforms) and class IIa (HDAC4, 5, 7, and 9 isoforms) HDACs, but not class IIb (HDAC6 and 10 isoforms)90, 91. This results in histone hyperacetylation and a more relaxed chromatin conformation. Enhanced access to specific gene promoters facilitates transcription factor binding and regulates the expression of numerous genes, notably neurotrophic and neuroprotective factors such as BDNF and Bcl-214.
Anti-excitotoxic effects
VPA can block glutamate-induced excitotoxicity in cultured neurons. By inhibiting HDACs, VPA upregulates α-synuclein, which suppresses glutamate neurotoxicity in rat cerebellar granule cells92. In addition, VPA has been shown to protect mature cerebellar granule cells from NMDA receptor-mediated excitotoxicity induced by SYM 2081, an inhibitor of excitatory amino acid transporters and an agonist of low-affinity kainate receptors93. Another study noted that treatment with lithium or VPA inhibited glutamate-induced excitotoxicity by inhibiting oxidative stress in rat cerebral cortical neurons, as shown by reduced lipid peroxidation and protein oxidation94. In addition, co-treatment with lithium and VPA completely blocked glutamate excitotoxicity in aging cerebellar granular cells, whereas treatment with lithium or VPA alone had no such neuroprotective effect95. Enhanced GSK-3 inhibition through serine phosphorylation is most likely a molecular target for this synergy95.
Anti-apoptotic effects
Anti-apoptotic signaling is also involved in VPA's neuroprotective effects against both ischemic and hemorrhagic stroke. In transient and permanent focal cerebral ischemic rat models, post-insult VPA treatment restored the loss of histone acetylation, superinduced HSP70, and suppressed phosphorylated Akt downregulation, caspase-3 activation and p53 overexpression following injury87, 88. Similarly, two distinct HDAC inhibitors, sodium butyrate (SB) and hydroxamic acid trichostatin A (TSA) were found to have anti-apoptotic effects in a permanent cerebral ischemic model88. The neuroprotective time window for reducing infarct volume and improving neurological performance was approximately three to six hours after ischemic onset for VPA and SB. VPA also exerted anti-apoptotic actions in a rat intracerebral hemorrhagic model of stroke. Here, VPA upregulated Bcl-2/Bcl-xl, downregulated Bax, inhibited caspase activity, and reduced the number of apoptotic TUNEL-positive cells96. In this study, VPA also enhanced protein levels of HSP70, phosphorylation of ERK, Akt and CREB, as well as acetylation of histone-H3. Together, these data suggest that HDAC inhibition is involved in VPA's anti-apoptotic effects.
Anti-inflammatory effects
VPA was first shown to display robust anti-inflammatory effects in midbrain neuron-glia co-cultures stimulated with LPS, as evidenced by the suppression of LPS-induced TNF-α secretion and nitric oxide production97, 98. In a rat permanent ischemic model of stroke, post-insult treatment with 300 mg/kg (ip) of VPA or SB strongly suppressed ischemia-induced microglial activation, reduced the number of infiltrating monocytes/macrophages, and inhibited overexpression of the proinflammatory enzymes iNOS and cyclooxygenase-2 (COX-2)88. In addition, HDAC inhibitors were found to superinduce HSP70, preserve levels of phosphorylated Akt, and inhibit ischemia-induced overexpression of p53 in the ischemic brain87, 88, 99. VPA-induced HSP70 is triggered by acetylation and recruitment of the transcription factor Sp1 to the HSP70 promoter via inhibition of Class I HDACs100. Recent work demonstrated that histone 3 lysine 4 methylation levels at the HSP70 promoter were enhanced by VPA-induced HDAC inhibition in both neurons and astrocytes101. A study in a mouse MCAO model found that HSP70 overexpression inactivated the key inflammatory transcription factor nuclear factor-κB (NF-κB) by stabilizing the NF-κB-IκB complex, thus preventing nuclear translocation of activated NF-κB subunits102. Furthermore, VPA and SB significantly inhibited MCAO-induced NF-κB activation in a transient MCAO rat model103. Hence, it is likely that superinduction of endogenous HSP70 by HDAC inhibition contributes to the anti-inflammatory effects of VPA via NF-κB inhibition.
VPA also exerted anti-inflammatory effects in an intracerebral hemorrhagic model of stroke96. Post-insult VPA treatment alleviated cerebral inflammation by inhibiting neutrophil infiltration, suppressing microglial activation, and downregulating mRNA levels of pro-inflammatory cytokines such as Fas ligand and IL-696. VPA treatment markedly upregulated acetylated histone H3 levels in both cerebral ischemic and hemorrhagic models88, 96, 103. These findings strongly support the anti-inflammatory effects of HDAC inhibition.
Blood-brain barrier protection
A recent study from our laboratory reported that post-insult treatment with VPA (200 and 300 mg/kg, ip) attenuated BBB disruption and brain edema in a rat model of transient focal cerebral ischemia103. VPA-induced BBB protection was dose-dependent and persisted for at least three days after cerebral ischemia and reperfusion. Notably, VPA administered at the beginning of reperfusion following a 60-min MCAO also significantly protected BBB integrity, although this effect was not as robust as that seen when VPA was given immediately after occlusion103. By restoring histone acetylation, VPA strongly inhibited cerebral ischemia-induced nuclear translocation of NF-κB, upregulation of MMP-9, degradation of tight junctions, and brain edema103. Moreover, the HDAC inhibitor SB was similarly found to have protective effects against BBB disruption, NF-κB activation, and MMP-9 upregulation103. It thus appears that in ischemic animal models, the BBB protection by VPA likely involves the initial inhibition of HDACs followed by suppression of NF-κB activation and MMP-9 overexpression, resulting in overall beneficial effects.
Pro-angiogenic effects
Recent work from our laboratory found that chronic post-ischemic VPA treatment enhanced post-ischemic angiogenesis, reduced brain infarction, and improved rotarod performance in an experimental ischemic stroke model (unpublished results). Chronic VPA treatment increased microvascular endothelial cell proliferation and enhanced the relative cerebral blood flow in the penumbra cortex, as detected by perfusion-weighted MRI. The pro-angiogenic effects of VPA may be attributable to HDAC inhibition-triggered upregulation of VEGF and MMP-9. Together, these findings lend further support to the notion that mood stabilizers promote post-ischemic angiogenesis in experimental stroke models, and suggest that these pro-angiogenic effects may contribute to their beneficial effects on long-term functional recovery after stroke.
Pro-neurogenic effects
VPA was found to promote cortical neurogenesis in primary cultures and to enhance hippocampal neurogenesis in adult mice by activating the ERK1/2 pathway104. The initial mechanism by which VPA activates the ERK pathway is unlikely to be mediated solely by HDAC inhibition, as its effects are not fully mimicked by TSA, another HDAC inhibitor104. However, it has been shown that treatment with the HDAC inhibitors VPA, SB or TSA largely promoted neuronal differentiation in cultured hippocampal neural progenitor cells105. Therefore, the ERK pathway is not exclusively responsible for the neuronal differentiation observed in neural progenitor cells.
Chronic treatment with the HDAC inhibitors SB (300 mg/kg, sc) and TSA (0.2 mg/kg, sc) was also found to significantly enhance post-ischemic cell proliferation and neurogenesis in a rat model of permanent cerebral ischemia. This occurred not only in the SVZ and SGZ but also in the ischemic cortex and striatum106. SB treatment upregulated levels of BDNF, phospho-CREB and GFAP in multiple brain regions after ischemia 106. In addition, intracerebral injection of K252a, a TrkB tyrosine kinase inhibitor, blocked SB-induced cell proliferation, neuronal differentiation and CREB activation in multiple brain regions, in addition to blocking the overall behavioral benefits of this agent106. These findings suggest that HDAC inhibitor-induced cell proliferation, neuronal differentiation and migration require BDNF-TrkB signaling, which may contribute to the long-term beneficial effects on behavioral performance observed after ischemic injury. Notably, lithium, through GSK-3 inhibition, and VPA, by HDAC inhibition, have both been shown to selectively activate BDNF promoter IV activity in rat cortical neurons107.
Effects on stem cell migration
For the past decade, stem cell therapy has been investigated as a potential treatment for stroke. Mesenchymal stem cells (MSCs) derived from bone marrow have been employed in experimental stroke models and shown to improve the functional recovery of neurological deficits induced by cerebral ischemia108. However, it is increasingly recognized that the poor homing and migratory abilities of transplanted MSCs limit the effectiveness of this treatment strategy. Our recent in vitro findings demonstrated that treatment with lithium or VPA enhanced MSC migration by elevating MMP-9 levels through GSK-3β inhibition or by increasing CXC chemokine receptor 4 (CXCR4) via HDAC inhibition, respectively109. Furthermore, combined treatment with lithium and VPA was found to have additive effects on MSC migration109. In a follow-up in vivo study, MSCs were primed with lithium and/or VPA and then transplanted into transient focal cerebral ischemic rats at 24 h after ischemic onset110. Lithium or VPA priming significantly increased the number of MSCs homing to brain infarct regions two weeks after transplantation. Ischemic rats receiving lithium- and/or VPA-primed MSCs showed improved functional recovery, reduced infarct volume, and enhanced angiogenesis in the penumbra regions. Notably, MSCs co-primed with lithium and VPA showed remarkable improvement in homing ability and contributed robustly to functional recovery after transplantation into ischemic rats. These beneficial effects of lithium and VPA priming were reversed by pharmacological inhibition of MMP-9 and CXCR4, respectively, suggesting that the underlying mechanisms likely involved lithium-induced MMP-9 upregulation and VPA-induced CXCR4 overexpression. Together, these findings suggest the potential utility of priming MSCs with GSK-3 and HDAC inhibitors to enhance the migration and homing capacity after transplantation into stroke victims.
Lamotrigine
Therapeutic benefits
Lamotrigine has been demonstrated to exert neuroprotective actions in focal and global cerebral ischemic models. In a permanent MCAO rat model, post-insult treatment with this drug attenuated the neurological deficit and reduced the infarct volume at 24 h after MCAO at a narrow dose range between 8 and 20 mg/kg111. Lamotrigine at 20 mg/kg still showed protective effects when given one hour after MCAO. Lamotrigine also provided significant histological and behavioral protection in a gerbil global ischemic model112, 113. Lamotrigine treatment completely preserved hippocampal CA3 cell loss and greatly attenuated damage to CA1 and CA3 cells. These effects contributed to its behavioral benefits against ischemia-induced cognitive deficits113. Cerebral vasospasm occurs a few days after subarachnoid hemorrhage and results in ischemic neurologic deficits. It has also been shown that oral administration of lamotrigine had marked neuroprotective effects and significantly attenuated cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits114.
Primary target: voltage-gated sodium channels
Lamotrigine blocks voltage-gated sodium channels (VGSCs), which are key mediators of intrinsic neuronal and muscle excitability. A growing body of evidence implicates abnormal VGSC activity as central to the pathophysiology of epileptic seizures as well as the pathophysiology of diverse neurological disorders, including stroke15. Ischemia-induced energy depletion leads to decreased Na+/K+-ATPase pump activity, membrane depolarization, persistent inward Na+ currents, and abnormal intracellular accumulation of Na+. An overload of Na+ drives the Na+/Ca2+ exchanger to import Ca2+, triggers glutamate release, and causes further neuronal damage15. Hence, VGSC blockers such as lamotrigine might be effective in preventing neuronal injuries caused by decreased oxygen supply. Lamotrigine has been shown to mitigate neuronal excitability by blocking VGSCs, inhibiting presynaptic release of glutamate, and affecting downstream intracellular signaling pathways115, 116.
Anti-excitotoxic effects
Na+ influx following cerebral ischemia partially contributes to excessive glutamate release. As a VGSC blocker, lamotrigine may act at these channels to stabilize neuronal membranes, inhibit glutamate transmitter release, and protect the brain from excitotoxic injury115. Lamotrigine effectively inhibited extracellular glutamate accumulation after transient global cerebral ischemia in rabbits117. In addition, selective neuronal damage in the hippocampal CA1 region of gerbils occurred two to three days after transient cerebral ischemia118. Lamotrigine treatment attenuated this injury by preventing the ischemia-induced increase in post-synaptic intrinsic excitability of CA1 pyramidal neurons, thus mitigating glutamate excitotoxicity and cell loss119. In addition, a microdialysis study found a significant reduction in the ischemia-induced glutamate surge in lamotrigine-treated ischemic animals112. In a rat model of neonatal hypoxic-ischemic encephalopathy, lamotrigine also reduced the hippocampal tissue levels of glutamate and aspartate, but did not affect GABA or glutamine levels120. Taken together, these findings support the notion that the neuroprotective effects of lamotrigine against ischemic injury are at least partially mediated by a reduction in excitatory amino acids.
Anti-apoptotic effects
The anti-apoptotic effects of lamotrigine in stroke treatment were reported in a rat model of neonatal hypoxia-ischemia. In this study, lamotrigine protected neurons from necrosis and apoptosis, as indicated by significantly fewer TUNEL-positive cells in the cortex and dentate gyrus of the ischemic hemisphere following lamotrigine administration121. In human neuroblastoma cells, chronic treatment with lamotrigine, like lithium and VPA, protected against GSK-3β-facilitated apoptosis, as measured by reduced caspase-3 activity122. Therefore, it appears that the GSK-3β signaling system might be involved in the protective effects of multiple mood stabilizers. Furthermore, chronic lamotrigine administration increased Bcl-2 mRNA and protein levels in the frontal cortex of rats, suggesting that upregulation of this anti-apoptotic factor may contribute to its protective effects123.
Effects on neurogenesis
After seven consecutive daily injections in 21-day postnatal rats, lamotrigine, but not VPA, increased the number of BrdU-labeled cells in the granule cell layer of the dentate gyrus, suggesting increased neurogenesis124. In cultured adult dentate gyrus-derived neural precursor cells, lithium, VPA and lamotrigine decreased staurosporine-induced apoptosis81. VPA also reversed the decrease in proliferation induced by dexamethasone, while lamotrigine had no such effect. However, unlike lithium, VPA and lamotrigine elevated the ratio of astroglial differentiation to neuronal differentiation under retinoic acid stimulation81. The reason(s) for the different mechanisms of action of these three mood stabilizers requires further investigation.
Conclusions and future directions
In this review, we have discussed recent preclinical advances in understanding the neuroprotective effects of the mood stabilizers lithium, VPA and lamotrigine in experimental stroke models. Accumulating evidence strongly supports the notion that these mood stabilizers exert multiple effects aimed at diverse aspects of stroke pathophysiology, leading to significant protection following insult.
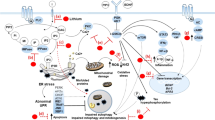
GSK-3 and HDACs are the primary targets of lithium and VPA, respectively, and initiate transcriptional activation of abundant downstream molecules in the ischemic brain, including neuroprotective and neurotrophic gene products, as illustrated in Figure 2. In line with this notion, several other pharmacological GSK-3 inhibitors125, 126, 127, 128 and HDAC inhibitors14 have been shown to exert beneficial effects against cerebral ischemia. Bcl-2 is transcriptionally activated by lithium through direct or indirect inhibition of GSK-3, while VPA induces Bcl-2 by direct HDAC inhibition. HSP70 expression can be enhanced by lithium-induced HSF-1 and VPA-induced Sp1 acetylation to enhance its promoter activity. Both Bcl-2 and HSP70 play prominent protective roles against glutamate excitotoxicity following stroke. HSP70 is likely a key player in the anti-inflammatory effects of VPA by inhibiting ischemia-induced NF-κB activation. The latter action may be involved in VPA's suppression of ischemia-induced short-term upregulation of MMP-9, which contributes to BBB disruption. Delayed increases in MMP-9 and VEGF induced by VPA are likely mediators of angiogenesis. BDNF can also be induced by lithium and VPA through their respective inhibition of GSK-3 and HDACs, and the BDNF signaling pathway is critical for HDAC inhibition-induced neurogenesis and functional improvement. In addition, lithium inhibits the NMDA receptor-mediated Ca2+ influx, presumably by decreasing receptor tyrosine phosphorylation. This, in turn, decreases excitotoxicity-induced activation of calpain, as well as p38 and JNK and downstream AP-1 signaling, thus protecting neurons from apoptosis. Lamotrigine similarly protects against ischemia-induced excitotoxicity, but this likely involves VGSC inhibition, which decreases synaptic glutamate accumulation and receptor overexcitation (Figure 2). It remains to be explored whether additional targets are involved in lamotrigine's neuroprotective effects. Since the direct targets that lithium, VPA and lamotrigine engage to elicit beneficial effects in experimental stroke models have recently become clear, further preclinical and clinical stroke studies using other specific agents that specifically inhibit these targets are suggested.

An overview of the proposed signaling mechanisms underlying the protective effects of mood stabilizing drugs against experimental stroke. Lithium, VPA and lamotrigine inhibit glutamate excitotoxicity-induced neuronal apoptosis by different mechanisms. Lithium inhibits stroke-induced NMDA receptor overactivation by decreasing NR2 subunit tyrosine phosphorylation. This suppresses excitotoxicity-induced activation of either calpain or p38, JNK and subsequently AP-1 to block neuronal apoptosis. VPA suppresses glutamate excitotoxicity via regulation of gene expression, among other mechanisms. As a VGSC blocker, lamotrigine attenuates stroke-induced VGSC malfunction, prevents extracellular glutamate overflow, and consequently mitigates excitotoxicity and apoptosis. Lithium and VPA inhibit GSK-3 and HDACs, respectively, to transcriptionally regulate various downstream neuroprotective and neurotrophic factors, as well as to enhance mesenchymal stem cell migration. Bcl-2, an antiapoptotic factor, is upregulated by lithium and VPA under experimental stroke conditions. Expression of the neuroprotective molecule HSP70 is enhanced by lithium and VPA through HSF-1 and Sp1 activation, respectively. HSP70 is also proposed to exert anti-inflammatory effects by inhibiting NF-κB activity. VPA attenuates BBB disruption by downregulating MMP-9 via NF-κB inhibition and preventing tight junction protein degradation shortly after ischemia. Long-term VPA treatment potentiates post-ischemic angiogenesis through upregulating VEGF and MMP-9 expression. Lithium also enhances VEGF and MMP-9 expression following long-term treatment. BDNF is activated by lithium and VPA, and the BDNF-TrkB signaling pathway is essential for enhancing post-ischemic neurogenesis and functional recovery. HAT: histone acetyltransferase; TJ: tight junctions; lines with solid arrows represent stimulatory effects; lines with flattened ends represent inhibitory effects; A: acetylated Lys residues of histone-tail proteins.
Compelling evidence supports the notion that HDAC inhibition by VPA and other compounds elicits strong anti-inflammatory effects in experimental stroke models. GSK-3β inhibition has been demonstrated to have anti-inflammatory effects, as shown by reduced TNF-α production via attenuated activation of NF-κB and JNK signaling cascades129, and induction of the anti-inflammatory cytokine IL-10130. In light of these findings, the anti-inflammatory effects of lithium in stroke conditions are recommended for future investigation. Emerging evidence suggests that specific microRNAs are regulated by mood stabilizers, and are involved in the translational suppression of multiple targets with major neurophysiological functions131. MicroRNAs have already been implicated in the pathogenesis of stroke132, 133; thus, further investigation along these lines will not only provide new insights into the neuroprotective effects of mood stabilizers, but also determine the potential utility of targeting specific microRNAs for stroke therapy.
Taken together, the promising preclinical evidence reviewed here, combined with these agents' long history of safe use in humans, suggests that long-term and large-scale clinical trials of mood stabilizers in stroke management are warranted.
References
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics−2011 update: a report from the American Heart Association. Circulation 2011; 123: e18–e209.
Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med 1995; 333: 1581–7.
Del Zoppo GJ, Saver JL, Jauch EC, Adams HP Jr . Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke 2009; 40: 2945–8.
Barber PA, Zhang J, Demchuk AM, Hill MD, Buchan AM . Why are stroke patients excluded from TPA therapy? An analysis of patient eligibility. Neurology 2001; 56: 1015–20.
Kleindorfer D, Lindsell CJ, Brass L, Koroshetz W, Broderick JP . National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke 2008; 39: 924–8.
Bearden CE, Woogen M, Glahn DC . Neurocognitive and neuroimaging predictors of clinical outcome in bipolar disorder. Curr Psychiatry Rep 2010; 12: 499–504.
Chuang DM . Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol 2004; 16: 83–90.
Chuang DM . The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci 2005; 1053: 195–204.
Manji HK, Drevets WC, Charney DS . The cellular neurobiology of depression. Nat Med 2001; 7: 541–7.
Chuang DM, Manji HK . In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psychiatry 2007; 62: 4–6.
Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK . Lithium-induced increase in human brain grey matter. Lancet 2000; 356: 1241–2.
Atmaca M, Ozdemir H, Cetinkaya S, Parmaksiz S, Belli H, Poyraz AK, et al. Cingulate gyrus volumetry in drug free bipolar patients and patients treated with valproate or valproate and quetiapine. J Psychiatr Res 2007; 41: 821–7.
Chiu CT, Chuang DM . Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol Ther 2010; 128: 281–304.
Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT . Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 2009; 32: 591–601.
Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M . Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol 2010; 9: 413–24.
Deb P, Sharma S, Hassan KM . Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 2010; 17: 197–218.
Allen CL, Bayraktutan U . Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 2009; 4: 461–70.
Ito U, Ohno K, Nakamura R, Suganuma F, Inaba Y . Brain edema during ischemia and after restoration of blood flow. Measurement of water, sodium, potassium content and plasma protein permeability. Stroke 1979; 10: 542–7.
Iadecola C, Alexander M . Cerebral ischemia and inflammation. Curr Opin Neurol 2001; 14: 89–94.
Wang Q, Tang XN, Yenari MA . The inflammatory response in stroke. J Neuroimmunol 2007; 184: 53–68.
Danton GH, Dietrich WD . Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 2003; 62: 127–36.
Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ . Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am J Pathol 1994; 144: 188–99.
Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R . Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2003; 183: 25–33.
Zhang Z, Chopp M, Powers C . Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res 1997; 744: 189–98.
Dong Y, Benveniste EN . Immune function of astrocytes. Glia 2001; 36: 180–90.
Sandoval KE, Witt KA . Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008; 32: 200–19.
Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov 2007; 6: 650–61.
Rosell A, Lo EH . Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol 2008; 8: 82–9.
Rosenberg GA, Navratil M, Barone F, Feuerstein G . Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab 1996; 16: 360–6.
Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH . Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res 1999; 842: 92–100.
Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ . Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab 1999; 19: 624–33.
Park KP, Rosell A, Foerch C, Xing C, Kim WJ, Lee S, et al. Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke 2009; 40: 2836–42.
Barr TL, Latour LL, Lee KY, Schaewe TJ, Luby M, Chang GS, et al. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke 2010; 41: e123–8.
Rosell A, Cuadrado E, Ortega-Aznar A, Hernandez-Guillamon M, Lo EH, Montaner J . MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008; 39: 1121–6.
Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci 2001; 21: 7724–32.
Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA . Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 2007; 27: 697–709.
Lo EH . A new penumbra: transitioning from injury into repair after stroke. Nat Med 2008; 14: 497–500.
Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K, et al. Cell-cell signaling in the neurovascular unit. Neurochem Res 2007; 32: 2032–45.
Jin K, Sun Y, Xie L, Childs J, Mao XO, Greenberg DA . Post-ischemic administration of heparin-binding epidermal growth factor-like growth factor (HB-EGF) reduces infarct size and modifies neurogenesis after focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 2004; 24: 399–408.
Chen J, Zhang ZG, Li Y, Wang Y, Wang L, Jiang H, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol 2003; 53: 743–51.
Wang L, Zhang Z, Wang Y, Zhang R, Chopp M . Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004; 35: 1732–7.
Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest 2000; 106: 829–38.
Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A . Targets for vascular protection after acute ischemic stroke. Stroke 2004; 35: 2220–5.
Hansen TM, Moss AJ, Brindle NP . Vascular endothelial growth factor and angiopoietins in neurovascular regeneration and protection following stroke. Curr Neurovasc Res 2008; 5: 236–45.
Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML . Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol 2005; 169: 681–91.
Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med 2006; 12: 441–5.
Nonaka S, Chuang DM . Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport 1998; 9: 2081–4.
Bian Q, Shi T, Chuang DM, Qian Y . Lithium reduces ischemia-induced hippocampal CA1 damage and behavioral deficits in gerbils. Brain Res 2007; 1184: 270–6.
Ren M, Senatorov VV, Chen RW, Chuang DM . Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci U S A 2003; 100: 6210–5.
Klein PS, Melton DA . A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 1996; 93: 8455–9.
Stambolic V, Ruel L, Woodgett JR . Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 1996; 6: 1664–8.
Jope RS . Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci 2003; 24: 441–3.
Ryves WJ, Harwood AJ . Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun 2001; 280: 720–5.
Chuang DM, Wang Z, Chiu CT . GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci 2011; 4: 15.
Ozaki N, Chuang DM . Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain. J Neurochem 1997; 69: 2336–44.
Chen RW, Qin ZH, Ren M, Kanai H, Chalecka-Franaszek E, Leeds P, et al. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. J Neurochem 2003; 84: 566–75.
Nonaka S, Hough CJ, Chuang DM . Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci U S A 1998; 95: 2642–7.
Takagi N, Shinno K, Teves L, Bissoon N, Wallace MC, Gurd JW . Transient ischemia differentially increases tyrosine phosphorylation of NMDA receptor subunits 2A and 2B. J Neurochem 1997; 69: 1060–5.
Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM . Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem 2002; 80: 589–97.
Ma J, Zhang GY . Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci Lett 2003; 348: 185–9.
Chalecka-Franaszek E, Chuang DM . Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A 1999; 96: 8745–50.
Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM . Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 2002; 43: 1173–9.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91: 231–41.
Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3K/Akt-dependent pathway. EMBO J 1997; 16: 6151–61.
Chen RW, Chuang DM . Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem 1999; 274: 6039–42.
Broughton BR, Reutens DC, Sobey CG . Apoptotic mechanisms after cerebral ischemia. Stroke 2009; 40: e331–9.
Mielke K, Herdegen T . JNK and p38 stresskinases--degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol 2000; 61: 45–60.
Whitmarsh AJ, Davis RJ . Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med 1996; 74: 589–607.
Xu J, Culman J, Blume A, Brecht S, Gohlke P . Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke 2003; 34: 1287–92.
Camins A, Crespo-Biel N, Junyent F, Verdaguer E, Canudas AM, Pallas M . Calpains as a target for therapy of neurodegenerative diseases: putative role of lithium. Curr Drug Metab 2009; 10: 433–47.
Li Q, Li H, Roughton K, Wang X, Kroemer G, Blomgren K, et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis 2010; 1: e56.
Bijur GN, Jope RS . Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3beta in the regulation of HSF-1 activity. J Neurochem 2000; 75: 2401–8.
Klionsky DJ, Emr SD . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–21.
Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008; 4: 762–9.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170: 1101–11.
Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005; 1: 11–22.
Li H, Li Q, Du X, Sun Y, Wang X, Kroemer G, et al. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J Cereb Blood Flow Metab 2011; 31: 2106–15.
Beurel E, Jope RS . Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation 2009; 6: 9.
Kim YR, van Meer MP, Tejima E, Murata Y, Mandeville JB, Dai G, et al. Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke 2008; 39: 439–47.
Guo S, Arai K, Stins MF, Chuang DM, Lo EH . Lithium upregulates vascular endothelial growth factor in brain endothelial cells and astrocytes. Stroke 2009; 40: 652–5.
Boku S, Nakagawa S, Masuda T, Nishikawa H, Kato A, Toda H, et al. Effects of mood stabilizers on adult dentate gyrus-derived neural precursor cells. Prog Neuropsychopharmacol Biol Psychiatry 2011; 35: 111–7.
Kim JS, Chang MY, Yu IT, Kim JH, Lee SH, Lee YS, et al. Lithium selectively increases neuronal differentiation of hippocampal neural progenitor cells both in vitro and in vivo. J Neurochem 2004; 89: 324–36.
Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK . Enhancement of hippocampal neurogenesis by lithium. J Neurochem 2000; 75: 1729–34.
Yan XB, Hou HL, Wu LM, Liu J, Zhou JN . Lithium regulates hippocampal neurogenesis by ERK pathway and facilitates recovery of spatial learning and memory in rats after transient global cerebral ischemia. Neuropharmacology 2007; 53: 487–95.
Shioda N, Han F, Fukunaga K . Role of Akt and ERK signaling in the neurogenesis following brain ischemia. Int Rev Neurobiol 2009; 85: 375–87.
Yoshida S, Kirino T, Tamura A, Basugi N, Sano K . Lithium ion does not protect brain against transient ischemia in gerbils. Stroke 1991; 22: 84–9.
Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM . Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem 2004; 89: 1358–67.
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM . Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther 2007; 321: 892–901.
Qian YR, Lee MJ, Hwang S, Kook JH, Kim JK, Bae CS . Neuroprotection by valproic Acid in mouse models of permanent and transient focal cerebral ischemia. Korean J Physiol Pharmacol 2010; 14: 435–40.
Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001; 20: 6969–78.
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS . Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001; 276: 36734–41.
Leng Y, Chuang DM . Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci 2006; 26: 7502–12.
Kanai H, Sawa A, Chen RW, Leeds P, Chuang DM . Valproic acid inhibits histone deacetylase activity and suppresses excitotoxicity-induced GAPDH nuclear accumulation and apoptotic death in neurons. Pharmacogenomics J 2004; 4: 336–44.
Shao L, Young LT, Wang JF . Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry 2005; 58: 879–84.
Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM . Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci 2008; 28: 2576–88.
Sinn DI, Kim SJ, Chu K, Jung KH, Lee ST, Song EC, et al. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis 2007; 26: 464–72.
Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, et al. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res 2005; 134: 162–9.
Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry 2006; 11: 1116–25.
Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, et al. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol 2006; 70: 1876–84.
Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, et al. Valproic acid induces functional heat-shock protein 70 via class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem 2009; 111: 976–87.
Marinova Z, Leng Y, Leeds P, Chuang DM . Histone deacetylase inhibition alters histone methylation associated with heat shock protein 70 promoter modifications in astrocytes and neurons. Neuropharmacology 2011; 60: 1109–15.
Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA . Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab 2008; 28: 53–63.
Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM . Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J Cereb Blood Flow Metab 2011; 31: 52–7.
Hao Y, Creson T, Zhang L, Li P, Du F, Yuan P, et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci 2004; 24: 6590–9.
Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH . Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A 2004; 101: 16659–64.
Kim HJ, Leeds P, Chuang DM . The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem 2009; 110: 1226–40.
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM . The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry 2009; 14: 51–9.
Borlongan CV . Bone marrow stem cell mobilization in stroke: a 'bonehead' may be good after all! Leukemia 2011. doi: 10.1038/leu.2011.167
Tsai LK, Leng Y, Wang Z, Leeds P, Chuang DM . The mood stabilizers valproic acid and lithium enhance mesenchymal stem cell migration via distinct mechanisms. Neuropsychopharmacology 2010; 35: 2225–37.
Tsai LK, Wang Z, Munasinghe J, Leng Y, Leeds P, Chuang DM . Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke 2011; 42: 2932–9.
Smith SE, Meldrum BS . Cerebroprotective effect of lamotrigine after focal ischemia in rats. Stroke 1995; 26: 117–21; discussion 21–2.
Shuaib A, Mahmood RH, Wishart T, Kanthan R, Murabit MA, Ijaz S, et al. Neuroprotective effects of lamotrigine in global ischemia in gerbils. A histological, in vivo microdialysis and behavioral study. Brain Res 1995; 702: 199–206.
Wiard RP, Dickerson MC, Beek O, Norton R, Cooper BR . Neuroprotective properties of the novel antiepileptic lamotrigine in a gerbil model of global cerebral ischemia. Stroke 1995; 26: 466–72.
Seckin H, Yigitkanli K, Besalti O, Kosemehmetoglu K, Ozturk E, Simsek S, et al. Lamotrigine attenuates cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. Surg Neurol 2008; 70: 344–51; discussion 51.
Leach MJ, Marden CM, Miller AA . Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II. Neurochemical studies on the mechanism of action. Epilepsia 1986; 27: 490–7.
Kuo CC, Lu L . Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol 1997; 121: 1231–8.
Bacher A, Zornow MH . Lamotrigine inhibits extracellular glutamate accumulation during transient global cerebral ischemia in rabbits. Anesthesiology 1997; 86: 459–63.
Kirino T . Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 1982; 239: 57–69.
Fan Y, Deng P, Wang YC, Lu HC, Xu ZC, Schulz PE . Transient cerebral ischemia increases CA1 pyramidal neuron excitability. Exp Neurol 2008; 212: 415–21.
Papazisis G, Kallaras K, Kaiki-Astara A, Pourzitaki C, Tzachanis D, Dagklis T, et al. Neuroprotection by lamotrigine in a rat model of neonatal hypoxic-ischaemic encephalopathy. Int J Neuropsychopharmacol 2008; 11: 321–9.
Yi YH, Guo WC, Sun WW, Su T, Lin H, Chen SQ, et al. Neuroprotection of lamotrigine on hypoxic-ischemic brain damage in neonatal rats: Relations to administration time and doses. Biologics 2008; 2: 339–44.
Li X, Bijur GN, Jope RS . Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord 2002; 4: 137–44.
Chang YC, Rapoport SI, Rao JS . Chronic administration of mood stabilizers upregulates BDNF and bcl-2 expression levels in rat frontal cortex. Neurochem Res 2009; 34: 536–41.
Kondziella D, Strandberg J, Lindquist C, Asztely F . Lamotrigine increases the number of BrdU-labeled cells in the rat hippocampus. Neuroreport 2011; 22: 97–100.
Collino M, Thiemermann C, Mastrocola R, Gallicchio M, Benetti E, Miglio G, et al. Treatment with the glycogen synthase kinase-3beta inhibitor, TDZD-8, affects transient cerebral ischemia/reperfusion injury in the rat hippocampus. Shock 2008; 30: 299–307.
Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, Luo J, et al. Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol 2004; 188: 378–86.
Koh SH, Yoo AR, Chang DI, Hwang SJ, Kim SH . Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem Biophys Res Commun 2008; 371: 894–9.
Valerio A, Bertolotti P, Delbarba A, Perego C, Dossena M, Ragni M, et al. Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J Neurochem 2011; 116: 1148–59.
Wang MJ, Huang HY, Chen WF, Chang HF, Kuo JS . Glycogen synthase kinase-3beta inactivation inhibits tumor necrosis factor-alpha production in microglia by modulating nuclear factor kappaB and MLK3/JNK signaling cascades. J Neuroinflammation 2010; 7: 99.
Huang WC, Lin YS, Wang CY, Tsai CC, Tseng HC, Chen CL, et al. Glycogen synthase kinase-3 negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cells. Immunology 2009; 128: e275–86.
Zhou R, Yuan P, Wang Y, Hunsberger JG, Elkahloun A, Wei Y, et al. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology 2009; 34: 1395–405.
Tan JR, Koo YX, Kaur P, Liu F, Armugam A, Wong PT, et al. microRNAs in stroke pathogenesis. Curr Mol Med 2011; 11: 76–92.
Jeyaseelan K, Lim KY, Armugam A . MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 2008; 39: 959–66.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Mental Health (NIMH), National Institutes of Health, and the Hsu family gift fund. The authors wish to thank Ioline Henter of the NIMH for her editorial assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Zf., Fessler, E. & Chuang, DM. Beneficial effects of mood stabilizers lithium, valproate and lamotrigine in experimental stroke models. Acta Pharmacol Sin 32, 1433–1445 (2011). https://doi.org/10.1038/aps.2011.140
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.140
Keywords
This article is cited by
-
Endothelial dysfunction in neuroprogressive disorders—causes and suggested treatments
BMC Medicine (2020)
-
Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of α-tubulin acetylation and FGF-21 up-regulation
Scientific Reports (2016)
-
Protection of Neurovascular Unit Cells with Lithium Chloride and Sodium Valproate Prevents Brain Damage in Neonatal Ischemia/Hypoxia
Bulletin of Experimental Biology and Medicine (2016)
-
Lithium Modulates Cancer Cell Growth, Apoptosis, Gene Expression and Cytokine Production in HL-60 Promyelocytic Leukaemia Cells and Their Drug-Resistant Sub-clones
Biological Trace Element Research (2012)
