
Abstract
The kidneys compose approximately 0.5 % of the body mass but consume about 10 % of the oxygen in cellular respiration. This discordance is due to the high energy demands on the kidney for reabsorption of filtered blood components and makes the kidney sensitive to stress on the mitochondria, the primary source of cellular ATP. Regardless of the etiology, acute kidney injury (AKI) almost always involves aspects of mitochondrial dysfunction. Recent evidence from experimental models suggests that preserving mitochondrial function or promoting mitochondrial repair rescues renal function during AKI. In this review we discuss the effect of AKI on disruption of mitochondrial homeostasis, and how the dynamic processes of mitochondrial biogenesis, fission/fusion, and mitophagy influence renal injury and recovery.
Similar content being viewed by others


Introduction
The primary functions of the kidney are to regulate body fluid volume, electrolyte balance, and excretion of waste products such as urea, ammonia, and xenobiotics. The kidney also secretes hormones (e.g., renin) which, along with maintaining fluid volume, help regulate blood pressure. There is a high demand for energy in the kidney due to the constant reabsorption of ions, small molecules, and peptides which occurs through active transport in the tubular epithelium. Renal epithelial cells, therefore, rely heavily on mitochondrial oxidative phosphorylation to supply sufficient ATP, which renders the kidney susceptible to injury due to energy deprivation and mitochondrial toxicants.
Acute kidney injury (AKI) is defined as an abrupt reduction in renal function associated with decreased urine output and an accumulation of serum waste products, namely urea and creatinine [1•] (Fig. 1). The prevalence of AKI is 2–7 % in hospital inpatients [2] and increases dramatically with diagnosis of sepsis [3] or admission to the ICU [4, 5]. AKI mortality is 50–80 % depending on severity of injury and population studied, and has not changed for decades [6–9]. Additionally, the medical expenses associated with treatment of AKI have been estimated to exceed $8 billion per year in the U.S. [10, 11]. Given the absence of viable drug therapies for AKI, it is clear that new targets are needed for therapeutic intervention. In this minireview we address recent advances in the field identifying mitochondrial dysfunction as a potential therapeutic target for AKI.

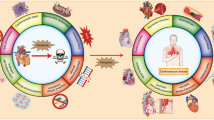
Dynamic processes affecting mitochondrial injury and recovery during the progression of AKI. Following insult to the kidney (ischemia, endotoxemia, toxicant exposure), development of AKI is defined as a loss of renal function, and characterized by a rise in serum creatinine. At the cellular level, elevated intracellular and mitochondrial calcium concentrations, increased production of reactive oxygen species (ROS), mitochondrial outer membrane permeabilization (MOMP), and loss of mitochondrial membrane potential (ΔΨ m) and ATP production, coincide with mitochondrial damage, fragmentation, and dysfunction. Over time, mitochondrial and renal function can be restored (blue line), and specific mitochondrial-targeted processes may facilitate recovery, including enhanced PGC-1α/mitochondrial biogenesis signaling [47••], stimulation of autophagy [82••, 85••], inhibition of Drp1/mitochondrial fission [64••, 65], or reduced Bax/Bak expression [72••]. Persistent mitochondrial and kidney injury (red line) may result under conditions of PGC-1α deficiency [48••], insufficient autophagy [79••, 81, 82••], or reduced Mfn2/mitochondrial fusion [66] (Color figure online)
Mitochondrial Dysfunction is a Cornerstone of AKI Pathophysiology
Mitochondrial damage is a major contributor to the lethal and sublethal tubular cell injury observed in the initiation and progression of AKI [12–16]. Increased production of reactive oxygen species (ROS), formed prominently within the mitochondria, as well as compromised antioxidant mechanisms after injury make the mitochondria particularly susceptible [17–20]. Additionally, elevations in intracellular and mitochondrial Ca2+ and Fe3+ may contribute to the central role of the mitochondria in the disease process [14, 21] (Fig. 1).
There is ample evidence indicating that mitochondrial dysfunction precedes renal dysfunction in animal models of AKI. Isolated proximal tubules (PT) subjected to hypoxia/reperfusion developed severe mitochondrial functional deficits, decreased mitochondrial membrane potential, and persistent ATP depletion [12, 15, 22]. In vivo, mitochondrial membrane potential decreased as early as 2 min after the onset of renal ischemia [23•]. Similar results were found using ex vivo imaging of kidney slices after 10 min of reperfusion [19]. The nephrotoxin HgCl2 inhibited mitochondrial ADP uptake and state 3 mitochondrial respiration in <3 h in vivo, while renal function was not affected until 12 h [24]. In animal models of septic AKI, treatment with lipopolysaccharide (LPS) decreased renal ATP [25] and increased renal lactate levels [26] in <4 h, well before the onset of AKI. Mitochondrial structure and function were damaged in the proximal and distal tubules 3 h after direct infusion of LPS to isolated kidneys [27]. These temporal studies suggest that mitochondrial dysfunction is an early pathology in AKI and may be driving the cell death and organ failure.
Persistent mitochondrial dysfunction is also a hallmark of AKI. Funk et al. [28] showed that several markers of mitochondrial content, including electron transport chain (ETC) proteins and mitochondrial DNA (mtDNA) copy number, decrease quickly after ischemia/reperfusion-(I/R) and myoglobinuric-AKI and remain suppressed for at least six days. In a gentamicin-induced AKI model, mitochondrial respiration defects and opening of the mitochondrial permeability transition pore (MPTP) were seen after six days [29]. Zsengeller et al. [30] used functional electron microscopy to demonstrate that cytochrome c oxidase (COX) enzyme activity decreased 3 days after a dose of the chemotherapeutic drug cisplatin that caused AKI at day 1. We suggest that mitochondrial dysfunction persists even with partial or complete recovery of renal function [28] and may be a sensitive biomarker of renal function.
The localization of mitochondrial dysfunction raises interesting questions about its impact on renal function. While the outer stripe of the outer medulla (OSOM) is the primary site of cell death in many AKI models including cisplatin [31], I/R [32], and HgCl2 [33], recent studies by Funk et al. and others, have characterized specific mitochondrial dysfunction in the renal cortex in the absence of gross cell death [28–30]. This observation indicates that (1) mitochondrial function is disrupted outside the primary target area (e.g., the S3 segment of the PT), (2) renal function is also decreased in the outer cortex, and (3) potential therapeutics also need to address this mitochondrial dysfunction.
There is extensive evidence of mitochondrial dysfunction in acute organ failure other than the kidney. Sepsis-induced heart failure caused myriad toxic effects on myocardial mitochondria including swelling, decreased respiration, and increased ROS production [34]. Liver failure in humans is most commonly caused by acetaminophen (APAP) [35], which causes hepatotoxicity through its toxic metabolite by induction of mitochondrial ROS [36]. Traumatic brain injury induced mitochondrial dysfunction includes increases in ROS production and reduced mitochondrial respiration rates [37, 38]. The significant connection between mitochondrial dysfunction and acute and critical diseases has been reviewed [39] and suggests that mitochondria are a potential therapeutic target for diseases including AKI, which currently have limited therapies. The remainder of this review will focus on specific pathways affecting mitochondrial homeostasis, particularly in AKI.
Mitochondrial Biogenesis
Mitochondrial biogenesis involves a complex, coordinated effort pairing a nuclear signal and response with a mitochondrial response. A variety of factors such as exercise, cold exposure, and injury, can stimulate mitochondrial biogenesis by turning on signaling pathways including NOS/cGMP, p38 MAPK, SIRT1, and AMPK [40]. The program includes activation of nuclear hormone receptors such as the peroxisome proliferator activated receptors (PPARs) and estrogen-related receptors (ERRs), and transcription factors and co-activators including PPARγ co-activator-1α (PGC-1α), the master regulator of mitochondrial biogenesis. The majority of genes expressed in mitochondria are transcribed from the nuclear genome, synthesized, and translocated to the mitochondrion because the mitochondrial genome encodes just 13 ETC proteins. The complete mitochondrial biogenic program has been comprehensively reviewed elsewhere [41].
Initial observations that PGC-1α and mitochondrial biogenesis may play a pivotal role in renal cell recovery were performed in primary renal proximal tubule cell (RPTC) culture models with oxidant injury [42–44]. Severe mitochondrial dysfunction occurred within 24 h of acute oxidant exposure, represented by dramatic reductions in ATP levels and mitochondrial oxygen consumption which spontaneously recovered over several days [43, 45]. Recovery of mitochondrial function in oxidant-injured RPTC was further clarified with the discovery that PGC-1α was robustly elevated in response to the insult via a Src-EGFR-p38 MAPK signaling pathway, and recovery could be expedited through adenoviral overexpression of PGC-1α or by pharmacological stimulation of mitochondrial biogenesis [42, 44, 46]. In a related study examining glomerular toxicity, Yuan et al. [47••] reported decreased PGC-1α expression, which correlated with mitochondrial and podocyte injury, following exposure to aldosterone. The authors also found that mitochondrial and cell injury could be attenuated by overexpressing PGC-1α or by overexpressing or pharmacologically stimulating SIRT1, a key regulator of PGC-1α activity.
The role of PGC-1α in recovery of renal function was recently explored in an endotoxic AKI model [48••]. Marked mitochondrial dysfunction was observed in the cortex and the corticomedullary junction of LPS-exposed mice, which was characterized by the loss of cytochrome c oxidase expression and activity, mitochondrial swelling, and rarefaction of mitochondrial cristae [48••]. Renal expression of PGC-1α mapped predominantly to cortical PTs, and expression correlated with kidney function, with PGC-1α mRNA and protein suppressed early after injury and restored during recovery [48••]. Finally, global and PT-specific PGC-1α knockout resulted in persistent AKI following endotoxin exposure, confirming that recovery of renal function was dependent on recovery of PGC-1α expression [48••] (Fig. 1).
In agreement with the report by Tran et al., recent studies from our laboratory examined mitochondrial dysfunction in the outer cortex of a renal I/R injury model, a region outside of the overt necrosis seen in the OSOM, and demonstrated suppressed PGC-1α mRNA early after reperfusion (between 0 and 72 h) which was restored as renal function partially recovered (between 72 and 144 h after reperfusion) [28]. Interestingly, PGC-1α protein was robustly expressed at the later time points in both the I/R model and glycerol-induced myoglobinuric injury model [28]; however, ETC proteins did not recover in the same time frame, which is consistent with the lack of full recovery of PT histology or renal function markers. Our unpublished observations revealed that restoration of mitochondrial proteins coincides with full recovery of renal function (approximately 2 weeks after injury), suggesting a delayed restoration following the induction of PGC-1α. In addition to the studies described, suppressed PGC-1α mRNA has also been reported in cisplatin nephrotoxicity, supporting the concept that signaling for mitochondrial biogenesis is decreased after injury and recovery of biogenesis could promote renal functional recovery [28, 47••, 49]. In addition to the studies described here, Weinberg [50] has summarized some of the recent findings identifying mitochondrial biogenesis as a potential target in renal disease.
The case for PGC-1α as an important molecular switch in organ dysfunction and repair is supported by the findings in acute injuries to extra-renal tissues. Alterations in PGC-1α expression and mitochondrial biogenesis have also been reported in models of myocardial infarction (MI), partial hepatectomy, and stroke [51–53], suggesting some overlapping mechanisms of injury and repair following mitochondrial-targeted injuries, although the exact responses may vary in different tissues. Reduced PGC-1α mRNA has been reported in heart failure in rats, and PGC-1α expression correlated with cardiac oxidative capacity, suggesting that reduced PGC-1α was involved in the pathophysiology of heart failure [51, 54]. The finding was confirmed in PGC-1α-deficient mice, which experienced accelerated cardiac dysfunction compared to matched wild type mice in a model of transverse aortic constriction [55]. More importantly, in studies examining gene expression in human cardiomyopathy, Sihag et al. [56] observed reduced PGC-1α and ERRα in failing hearts. The levels were correlated with left ventricular ejection fraction, which implicated reduced PGC-1α, ERRα, and mitochondrial biogenesis in the pathophysiology of heart failure. In an ischemic brain injury model, Yin et al. [53] reported an increase in mitochondrial biogenesis almost immediately after reperfusion. Although PGC-1α expression did not change, the authors observed increased mtDNA content, elevated mitochondrial proteins and citrate synthase activity, and increased expression of NRF-1 and mitochondrial transcription factor A (Tfam). In a related study, overexpression of Tfam was neuroprotective in a transient forebrain ischemia model [57]. As more data support the idea that mitochondrial biogenesis is a key target of acute organ failure, this pathway becomes a better target for therapeutic intervention to improve recovery.
Mitochondrial Dynamics
Mitochondrial morphology influences function, and morphological stability is governed by a balance of the dynamic processes controlling mitochondrial fusion and fission. Mitochondrial fusion, which involves multiple steps to tether membranes of adjacent mitochondria and share intramitochondrial components, is primarily driven by the dynamin-related GTPases, mitofusin1 (Mfn1) and mitofusin2 (Mfn2) at the outer mitochondrial membrane (OMM) and Opa1 on the inner membrane. Mitochondrial fission is primarily controlled by fission protein 1 (Fis1), which is localized on the OMM as an anchor protein for dynamin-related protein 1 (Drp1), a cytosolic GTPase recruited to mitochondrial scission sites. Additionally, recent findings have implicated the pro-apoptotic molecules, Bax and Bak, as regulators of mitochondrial morphology and fragmentation, intimately linked with mitochondrial outer membrane permeabilization (MOMP) and cell death [58, 59]. There are a number of constituents and biological factors that affect mitochondrial dynamics in various systems and disease states, which is beyond the scope of this review, but have been previously described in detail [60, 61, 62•].
Drp1 mRNA is highly expressed in the human kidney, in addition to the heart, brain, and skeletal muscle [63], and, therefore, may play a central role in the pathophysiology of mitochondrial-targeted injuries to these organs. Brooks et al. [64••] elegantly demonstrated the pathological consequences of excess mitochondrial fission after injury to renal cells and the acquired protection by blocking the response. The authors showed that both cisplatin and chemical anoxia induced Drp1-dependent mitochondrial fragmentation, which preceded cytochrome c release and apoptosis in RPTC. Dominant-negative Drp1 or siRNA-reduced expression of Drp1 preserved mitochondrial morphology, and protected against cytochrome c release and apoptosis in vitro [64••]. They further demonstrated mitochondrial fragmentation in I/R and cisplatin in in vivo models, and were able to protect renal function with mdivi-1, a pharmacological inhibitor of Drp1 [64••]. Similar renoprotective effects were observed with Drp1 inhibition in the glycerol-induced myoglobinuric model, in which mdivi-1 treatment attenuated tubular apoptosis compared to control animals [65] (Fig. 1).
A recent study examining kidney-specific Mfn2 deletion under basal and stressed conditions further supported the benefits of preserved mitochondrial fission/fusion balance following renal cell injury [66]. Mitochondrial fragmentation increased in kidneys and cultured renal epithelial cells from the Mfn2-knockout mice; however, oxygen consumption and ATP turnover were not affected, and apoptosis was not increased under basal conditions [66]. Following ATP depletion with sodium cyanide exposure, mitochondrial-associated Bax, cytosolic cytochrome c and apoptosis-inducing factor (AIF), and apoptotic cell death were elevated in the Mfn2-deficient cells compared to controls [66].
The Bcl-2 family proteins, Bax and Bak, have been implicated in human kidney disease [67–69], and although they are most commonly associated with progression of apoptosis, recent findings suggest a more complex role for these proteins in the maintenance of mitochondrial morphology under normal and stress conditions [62]. From a series of studies performed in his lab, Zheng Dong and colleagues described the involvement of Bak and Bax in mitochondrial fragmentation, mitochondrial permeabilization, and renal pathophysiology [70, 71, 72••]. Whereas it was associated with Mfn2 under physiological conditions, Bak associated with and inhibited Mfn1 under apoptotic conditions in several cell stress models [70]. Following azide, staurosporine, or cisplatin treatment, they observed mitochondrial fragmentation, cytochrome c release, and cell death, which was attenuated in Bak-deficient cells. Cytochrome c release and apoptosis, but not mitochondrial fragmentation, were also attenuated in Bax-deficient cells, illustrating the slightly different roles of these molecules in mitochondrial injury [70]. Importantly, in a murine model of renal I/R, Wei et al. [72••] demonstrated that global deletion of Bak inhibited mitochondrial fragmentation, suppressed cytochrome c release, and protected against tubule apoptosis and renal dysfunction. The authors also observed renal protection in mice with PT-specific Bax deletion, validating the function of these proteins as regulators of mitochondrial integrity and renal pathology in vivo [72••].
Outside of renal pathophysiology, disruption of mitochondrial dynamics appears to be a pivotal component of acute injuries to other tissues, including the heart and the brain. Ong et al. [73] demonstrated that inhibition of mitochondrial fission was cardioprotective in an ischemia–reperfusion injury. In cardiomyocyte cell models with simulated ischemia, the authors showed that overexpression of mitofusin proteins, Mfn1 or Mfn2, or inhibition of Drp1 was mitochondrial- and cyto-protective. Additionally, mdivi-1 pretreatment in a mouse model of MI significantly reduced infarct size compared to vehicle-treated animals at 2 h post-reperfusion [73]. Similarly, treatment with the Drp1 inhibitors mdiviA and mdiviB reduced infarct size in a mouse model of transient focal brain ischemia [74]. Additionally, and in agreement with findings in the kidney, Whelan et al. [75] reported preservation of mitochondrial morphology and cardioprotection in mice lacking Bak and Bax that underwent MI. The results of these studies suggest common mechanisms of mitochondrial membrane instability and permeability leading to organ dysfunction in various tissue injuries.
Autophagy and Mitophagy
Autophagy, also referred to as macroautophagy, is the bulk degradation of cytosolic components, including aged or damaged proteins, other macromolecules, and organelles. The process of autophagy involves the formation of a membranous structure, mediated by the initiating complex of class 3 phosphatidylinositol-3-kinase (PI3KCl3) and Beclin-1, the lipidated microtubule-associated protein light-chain-3 (LC3-II), and the adaptor protein p62, which sequesters flagged organelles or portions of cytosol into a vesicle known as an autophagosome. The autophagosome fuses with a lysosome to create an autolysosome, which proceeds to degrade the sequestered contents. In addition to the players listed above, a number of autophagy-specific proteins, identified as Atg proteins, have been characterized in yeast and mammalian systems [76]. Several of these are discussed in more detail below. Mitochondrial autophagy (mitophagy) is the selective engulfment of depolarized mitochondria, involving identification by PTEN-induced kinase 1 (PINK1) and ubiquitylation of OMM proteins by Parkin. The details surrounding PINK1- and Parkin-mediated mitophagy were extensively covered recently [77].
Several studies have examined the role of the autophagic response in AKI and renal disease since the finding that LC3 and Beclin-1 increased after I/R injury in the rat [78]. PT-specific Atg5 knockout mice accumulated morphologically-defective mitochondria and were more susceptible to I/R-induced PT apoptosis and elevated BUN and serum creatinine [79••]. Atg5 forms a complex with Atg12 and Atg16 that is essential in formation of the sequestration vesicle [80], and PT-specific Atg5 knockout mice were also more susceptible to cisplatin-induced AKI [81]. The authors found that cisplatin exposure exacerbated renal injury by functional markers and histopathology, and increased DNA damage, p53 activation, and apoptosis in PT-specific Atg5 knockout mice. Atg5-deficient PT cells exhibited an accumulation of ROS-producing mitochondria compared to wild type cells [81]. Jiang et al., also found that PT-specific deletion of Atg7, a key protein in the conjugation of phosphatidylethanolamine to LC3-I to form LC3-II, sensitized mice to cisplatin- and I/R-induced AKI [82••]. PT-Atg7 knockouts had a deficient autophagic response, elevated BUN and serum creatinine, and more severe tubular histopathology and apoptosis after AKI [82••]. The authors also demonstrated that pharmacological inhibition of autophagy produced results that were similar to those observed in the knockout studies, with elevated creatinine, BUN, and tissue damage in chloroquine-treated mice after cisplatin exposure [82••]. Although mitochondrial effects were not specifically described in the study, previous studies have demonstrated that loss of Atg7 function results in accumulation of defective mitochondria [83, 84]; therefore, it could be speculated that deficient mitochondrial autophagy contributed to the resultant phenotype. Conversely, promoting autophagy by pharmacological intervention may be renoprotective after AKI [82, 85••]. In a critically ill rabbit model, Gunst et al. [85••] identified insufficient autophagy in the kidneys and livers of hyperglycemic rabbits, which correlated with impaired mitochondrial function and, unlike mitochondrial fusion/fission and biogenesis, severity of organ dysfunction. By stimulating autophagy with rapamycin, the authors were able to improve renal mitochondrial complex V activity, and normalize plasma creatinine levels [85••] (Fig. 1).
In addition to the studies from experimental models, there is evidence that mitophagy may play a role in cellular repair in human renal disease. In postmortem analysis of kidneys taken from septic patients, Takasu et al. [86] described autophagosomes juxtaposed to membrane-damaged mitochondria in the tubular epithelium. The authors claimed that the autophagic response did not appear to be associated with cell death, but rather reversible injury. Autophagic vacuoles containing abnormal mitochondria were also reported in the kidneys of a hemolytic patient presenting with AKI, establishing a common mechanism for removal of damaged mitochondria in human renal failure [87].
Selective removal of damaged mitochondria has also been suggested as a protective mechanism in acute injuries to other tissues, including the heart and the liver. In a study examining the molecular mechanisms of mitophagy, it was found that cardiac mitophagy, which occurred after ischemic injury, was further promoted in p53-deficient mice, and corresponded with reduced accumulation of damaged mitochondria and cardioprotection [88]. The protective effects of p53 inhibition were reversed with chloroquine treatment, an inhibitor of autophagy. The protective effect of mitophagy was further illustrated in a model of cardiac ischemic preconditioning (IPC), as Parkin-deficient mice displayed dysfunctional p62 signaling and were resistant to IPC-cardioprotection [89]. Ni et al. [90] demonstrated the protective effects of autophagy in APAP hepatotoxicity. APAP induced autophagy in the livers of exposed mice, which included selective removal of damaged mitochondria, and inhibition of autophagy with chloroquine or 3-methyladenine exacerbated injury, while rapamycin-induced autophagy attenuated liver injury. Results from studies in the kidney and other organs indicate that autophagy, and specifically mitophagy, is a critical pathway for removal of damaged mitochondria and cellular repair after cell injury.
Conclusions
The preponderance of data supports a central role of mitochondrial dysfunction in the pathology of AKI. Numerous phenomenological and mechanistic studies have demonstrated that mitochondria are damaged at initiation of injury, and, at the very least, the recovery of mitochondrial function is correlated with recovery of renal function, if not a primary driver of recovery. As depicted in Fig. 1, mitochondria are at the center of a number of dynamic processes, initiated under either physiological or pathological conditions. Pathological signals leading to elevated levels of intracellular and mitochondrial Ca2+, increased production of ROS, mitochondrial membrane permeabilization and depolarization, and loss of ATP are hallmarks of mitochondrial dysfunction. Abnormal mitochondrial biogenesis, fission/fusion, and autophagy have been characterized in experimental models of AKI as well as in human kidney disease, and restoration of physiological levels may be a primary determinant of not only recovery of mitochondrial function, but also recovery of organ function. In light of the studies discussed in this review, a number of critical questions need to be addressed. (1) Are mitochondria a suitable target for therapeutic intervention in human AKI? (2) What cellular pathway(s) trigger mitochondrial dysfunction so consistently in renal injuries with distinct etiologies, and could these be pharmacologically manipulated? (3) Which mitochondrial repair pathway(s) are essential to recovery of organ function in human AKI? There is support from experimental models that recovery of the master regulator of mitochondrial biogenesis PGC-1α after AKI is necessary for recovery of renal function [47••, 48••]. Other studies have demonstrated the efficacy with drugs that target mitochondrial dynamics [64••, 65] and autophagy [82••, 85••]. While these agents will require more testing before being translated into clinical trials, we suggest mitochondria are a promising new target for a disease that has had no new drugs approved for therapy and has an unacceptably high mortality rate.
References
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
• Bellomo R, Kellum JA, Ronco C (2012) Acute kidney injury. Lancet 380 (9843): 756–766. Excellent review of clinical acute kidney injury
Nash K, Hafeez A, Hou S (2002) Hospital-acquired renal insufficiency. Am J Kidney Dis 39(5):930–936
Bagshaw SM (2006) The long-term outcome after acute renal failure. Curr Opin Crit Care 12(6):561–566
Bagshaw SM, George C, Bellomo R et al (2008) Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care 12(2):R47
Hoste EA, De Waele JJ (2008) Acute kidney injury in the intensive care unit: it’s the gene, stupid! Crit Care Med 36(12):3266–3267
Thadhani R, Pascual M, Bonventre JV (1996) Acute renal failure. N Engl J Med 334(22):1448–1460
Liano F, Junco E, Pascual J et al (1998) The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The Madrid Acute Renal Failure Study Group. Kidney Int Suppl 66:S16–S24
Schrier RW, Wang W, Poole B et al (2004) Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 114(1):5–14
VA/NIH Acute Renal Failure Trial Network, Palevsky PM, Zhang JH et al (2008) Intensity of renal support in critically ill patients with acute kidney injury. N Engl J Med 359(1):7–20
Hamel MB, Phillips RS, Davis RB et al (1997) Outcomes and cost-effectiveness of initiating dialysis and continuing aggressive care in seriously ill hospitalized adults. SUPPORT Investigators. Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments. Ann Intern Med 127(3):195–202
Fischer MJ, Brimhall BB, Lezotte DC et al (2005) Uncomplicated acute renal failure and hospital resource utilization: a retrospective multicenter analysis. Am J Kidney Dis 46(6):1049–1057
Weinberg JM, Venkatachalam MA, Roeser NF et al (2000) Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci USA 97(6):2826–2831
Jassem W, Fuggle SV, Rela M et al (2002) The role of mitochondria in ischemia/reperfusion injury. Transplantation 73(4):493–499
Jassem W, Heaton ND (2004) The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int 66(2):514–517
Feldkamp T, Kribben A, Weinberg JM (2005) Assessment of mitochondrial membrane potential in proximal tubules after hypoxia-reoxygenation. Am J Physiol 288(6):F1092–F1102
Hall AM, Unwin RJ (2007) The not so ‘mighty chondrion’: emergence of renal diseases due to mitochondrial dysfunction. Nephron Physiol 105(1):p1–p10
Shah SV, Walker PD (1988) Evidence suggesting a role for hydroxyl radical in glycerol-induced acute renal failure. Am J Physiol 255(3 Pt 2):F438–F443
Zager RA (1996) Mitochondrial free radical production induces lipid peroxidation during myohemoglobinuria. Kidney Int 49(3):741–751
Plotnikov EY, Kazachenko AV, Vyssokikh MY et al (2007) The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int 72(12):1493–1502
Plotnikov EY, Chupyrkina AA, Pevzner IB et al (2009) Myoglobin causes oxidative stress, increase of NO production and dysfunction of kidney’s mitochondria. Biochim Biophys Acta 1792(8):796–803
Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341(Pt 2):233–249
Weinberg JM, Venkatachalam MA, Roeser NF et al (2000) Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am J Physiol Renal Physiol 279(5):F927–F943
• Hall AM, Rhodes GJ, Sandoval RM et al (2013) In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int 83 (1): 72–83. The first study of mitochondrial function during AKI in live animals using intravital multiphoton microscopy
Weinberg JM, Harding PG, Humes HD (1982) Mitochondrial bioenergetics during the initiation of mercuric chloride-induced renal injury. II. Functional alterations of renal cortical mitochondria isolated after mercuric chloride treatment. J Biol Chem 257(1):68–74
Liaudet L, Fishman D, Markert M et al (1997) L-canavanine improves organ function and tissue adenosine triphosphate levels in rodent endotoxemia. Am J Respir Crit Care Med 155(5):1643–1648
Levy B, Mansart A, Bollaert PE et al (2003) Effects of epinephrine and norepinephrine on hemodynamics, oxidative metabolism, and organ energetics in endotoxemic rats. Intensive Care Med 29(2):292–300
Manny J, Livni N, Schiller M et al (1980) Structural changes in the perfused canine kidney exposed to the direct action of endotoxin. Isr J Med Sci 16(3):153–161
Funk JA, Schnellmann RG (2012) Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302(7):F853–F864
Morales AI, Detaille D, Prieto M et al (2010) Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int 77(10):861–869
Zsengeller ZK, Ellezian L, Brown D et al (2012) Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J Histochem Cytochem 60(7):521–529
Dobyan DC, Levi J, Jacobs C et al (1980) Mechanism of cis-platinum nephrotoxicity: II morphologic observations. J Pharmacol Exp Ther 213(3):551–556
Venkatachalam MA, Bernard DB, Donohoe JF et al (1978) Ischemic damage and repair in the rat proximal tubule: differences among the S1, S2, and S3 segments. Kidney Int 14(1):31–49
Gritzka TL, Trump BF (1968) Renal tubular lesions caused by mercuric chloride. Electron microscopic observations: degeneration of the pars recta. Am J Pathol 52(6):1225–1277
Smeding L, Plotz FB, Groeneveld AB et al (2012) Structural changes of the heart during severe sepsis or septic shock. Shock 37(5):449–456
Larson AM, Polson J, Fontana RJ et al (2005) Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42(6):1364–1372
Jaeschke H, McGill MR, Ramachandran A (2012) Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44(1):88–106
Verweij BH, Muizelaar JP, Vinas FC et al (2000) Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg 93(5):815–820
Lifshitz J, Sullivan PG, Hovda DA et al (2004) Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 4(5–6):705–713
Kozlov AV, Bahrami S, Calzia E et al (2011) Mitochondrial dysfunction and biogenesis: do ICU patients die from mitochondrial failure? Ann Intensive Care 1(1):41
Ventura-Clapier R, Garnier A, Veksler V (2008) Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79(2):208–217
Scarpulla RC, Vega RB, Kelly DP (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23(9):459–466
Rasbach KA, Schnellmann RG (2007) PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun 355(3):734–739
Rasbach KA, Schnellmann RG (2007) Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem 282(4):2355–2362
Rasbach KA, Funk JA, Jayavelu T et al (2010) 5-hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J Pharmacol Exp Ther 332(2):632–639
Nowak G, Aleo MD, Morgan JA et al (1998) Recovery of cellular functions following oxidant injury. Am J Physiol 274(3 Pt 2):F509–F515
Funk JA, Odejinmi S, Schnellmann RG (2010) SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther 333(2):593–601
•• Yuan Y, Chen Y, Zhang P et al (2012) Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic Biol Med 53 (1): 30–43. The authors found reduced PGC-1α expression following aldosterone exposure and glomerular toxicity. In the study, they demonstrated that by overexpressing PGC-1α or by overexpressing or pharmacologically-stimulating SIRT1 with resveratrol, podocytes were protected from aldosterone toxicity
•• Tran M, Tam D, Bardia A et al (2011) PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121 (10): 4003–4014. In this study, the authors demonstrated expression of genes associated with PGC-1α and oxidative phosphorylation were correlated with renal injury and recovery. Furthermore, in gene-deletion studies, it was revealed that PGC-1α expression was essential for recovery of renal function following endotoxin exposure
Portilla D, Dai G, McClure T et al (2002) Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int 62(4):1208–1218
Weinberg JM (2011) Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol 22(3):431–436
Rimbaud S, Garnier A, Ventura-Clapier R (2009) Mitochondrial biogenesis in cardiac pathophysiology. Pharmacol Rep 61(1):131–138
Wang H, Peiris TH, Mowery A et al (2008) CCAAT/enhancer binding protein-beta is a transcriptional regulator of peroxisome-proliferator-activated receptor-gamma coactivator-1alpha in the regenerating liver. Mol Endocrinol 22(7):1596–1605
Yin W, Signore AP, Iwai M et al (2008) Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 39(11):3057–3063
Garnier A, Fortin D, Delomenie C et al (2003) Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551(Pt 2):491–501
Arany Z, Novikov M, Chin S et al (2006) Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci USA 103(26):10086–10091
Sihag S, Cresci S, Li AY et al (2009) PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol 46(2):201–212
Hokari M, Kuroda S, Kinugawa S et al (2010) Overexpression of mitochondrial transcription factor A (TFAM) ameliorates delayed neuronal death due to transient forebrain ischemia in mice. Neuropathology 30(4):401–407
Karbowski M, Lee YJ, Gaume B et al (2002) Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159(6):931–938
Wasiak S, Zunino R, McBride HM (2007) Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177(3):439–450
Seo AY, Joseph AM, Dutta D et al (2010) New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci 123(Pt 15):2533–2542
Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89(3):799–845
• Zhan M, Brooks C, Liu F et al (2013) Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 83 (4): 568–581. Excellent review of mitochondrial dynamics in renal disease
Imoto M, Tachibana I, Urrutia R (1998) Identification and functional characterization of a novel human protein highly related to the yeast dynamin-like GTPase Vps1p. J Cell Sci 111(Pt 10):1341–1349
•• Brooks C, Wei Q, Cho SG et al (2009) Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119 (5): 1275–1285. In cell models of cisplatin toxicity and ATP depletion, Drp1 translocated to mitochondria prior to cytochrome c release and apoptosis, and could be blocked by dominant negative Drp1 or pharmacologic inhibition. Inhibition of mitochondrial fragmentation with mdivi-1 attenuated renal injury in rodent models of cisplatin nephrotoxicity and renal I/R injury
Tang WX, Wu WH, Qiu HY et al (2013) Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J Nephrol
Gall JM, Wang Z, Liesa M et al (2012) Role of mitofusin 2 in the renal stress response. PLoS One 7(1):e31074
Najarian JS et al (1994) The impact of the quality of initial graft function on cadaver kidney transplants. Transplantation 57(6):812–816
Castaneda MP et al (2003) Activation of mitochondrial apoptotic pathways in human renal allografts after ischemiareperfusion injury. Transplantation 76(1):50–54
Schwarz C et al (2002) Failure of BCL-2 up-regulation in proximal tubular epithelial cells of donor kidney biopsy specimens is associated with apoptosis and delayed graft function. Lab Invest 82(7):941–948
Brooks C, Wei Q, Feng L et al (2007) Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA 104(28):11649–11654
Brooks C, Dong Z (2007) Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle 6(24):3043–3047
•• Wei Q, Dong G, Chen JK et al (2013) Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. Global Bak and PT-specific Bax knockout mice were protected against I/R-induced AKI. Loss of these apoptotic proteins suppressed I/R-induced cytochrome c release and apoptosis but not necrosis
Ong SB, Subrayan S, Lim SY et al (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121(18):2012–2022
Grohm J, Kim SW, Mamrak U et al (2012) Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ 19(9):1446–1458
Whelan RS, Konstantinidis K, Wei AC et al (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA 109(17):6566–6571
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368(7):651–662
Jin SM, Youle RJ (2012) PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 125(Pt 4):795–799
Chien CT, Shyue SK, Lai MK (2007) Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 84(9):1183–1190
•• Kimura T, Takabatake Y, Takahashi A et al (2011) Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22 (5): 902–913. The authors observed an accumulation of abnormal mitochondria in proximal tubule cells from Atg5 −/− mice. Atg5 −/− mice were also more susceptible to I/R-induced apoptosis and renal dysfunction
Kuma A, Mizushima N, Ishihara N et al (2002) Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277(21):18619–18625
Takahashi A, Kimura T, Takabatake Y et al (2012) Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 180(2):517–525
•• Jiang M, Wei Q, Dong G et al (2012) Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82 (12): 1271–1283. Atg7 −/− mice were sensitized to I/R- and cisplatin-induced AKI, and rapamycin-stimulated autophagy attenuated renal injury
Komatsu M, Waguri S, Ueno T et al (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169(3):425–434
Wu JJ, Quijano C, Chen E et al (2009) Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 1(4):425–437
•• Gunst J, Derese I, Aertgeerts A et al (2013) Insufficient autophagy contributes to mitochondrial dysfunction, organ failure, and adverse outcome in an animal model of critical illness. Crit Care Med 41 (1): 182–194. Insufficient autophagy in the kidneys of hyperglycemic rabbits correlated with mitochondrial dysfunction and severity of organ injury. Rapamycin treatment promoted autophagy, improved mitochondrial function, and restored renal function
Takasu O, Gaut JP, Watanabe E et al (2013) Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med 187(5):509–517
Qian Q, Nath KA, Wu Y et al (2010) Hemolysis and acute kidney failure. Am J Kidney Dis 56(4):780–784
Hoshino A, Matoba S, Iwai-Kanai E et al (2012) p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol 52(1):175–184
Huang C, Andres AM, Ratliff EP et al (2011) Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 6(6):e20975
Ni HM, Bockus A, Boggess N et al (2012) Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55(1):222–232
Acknowledgments
This study was supported by the National Institutes of Health [R01 GM084147 to Rick G. Schnellmann, T32 HL007260 to L. Jay Stallons, T32 DK7752 to Jason A. Funk, and C06 RR015455], and by the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs. This publication was supported, in part, by the South Carolina Clinical and Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina CTSA, and funded by the National Institutes of Health [UL1 RR029882].
Disclosure
L. Jay Stallons, Jason A. Funk, and Rick G. Schnellmann declare that they have no conflict of interest.
Compliance with Ethics Guidelines
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stallons, L.J., Funk, J.A. & Schnellmann, R.G. Mitochondrial Homeostasis in Acute Organ Failure. Curr Pathobiol Rep 1, 169–177 (2013). https://doi.org/10.1007/s40139-013-0023-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-013-0023-x