
Abstract
Autophagy is the cell biology process in which cytoplasmic components are degraded in lysosomes to maintain cellular homeostasis and energy production. In the healthy kidney, autophagy plays an important role in the homeostasis and viability of renal cells such as podocytes and tubular epithelial cells and of immune cells. Recently, evidence is mounting that (dys)regulation of autophagy is implicated in the pathogenesis of various renal diseases, and might be an attractive target for new renoprotective therapies. In this review, we provide an overview of the role of autophagy in kidney physiology and kidney diseases.
Similar content being viewed by others

Autophagy: classification and interaction with apoptosis
Autophagy
Autophagy, Greek for “self-eating”, is the process in which cytoplasmic components are transported and delivered to lysosomes for degradation into their primary components, which can then be used as building blocks of novel essential macromolecules to maintain cellular homeostasis and energy production. It is different from heterophagy, in which cells degenerate extracellular substances. Recently, autophagy has been shown to be a critical player in normal human physiological processes. Defective autophagy is associated with a broad variety of pathological conditions, such as neurodegeneration, cardiomyopathy, and cancer, and with physiological responses to exercise and ageing [1, 2]. Indeed, autophagy is a major protective mechanism allowing cell survival in response to multiple stressors [3]. In general, baseline autophagy in mammalian systems occurs under normal conditions, but can be stimulated by starvation or by various pathologies including ischemic, toxic, immunological, and oxidative insults. Other autophagy stimulators are pharmacological agents like the macrolide antibiotic rapamycin, the best-known trigger for autophagy, which acts by inhibiting mechanistic/mammalian target of rapamycin (mTOR) [4].
Involvement of autophagy in renal diseases
Growing evidence suggests that autophagy activity is required for the homeostasis, viability, and physiological functions of renal cells and protects against age-related renal disease [5]. In experimental models of acute and chronic kidney diseases, autophagy has been shown to be crucial for renal cell survival. However, it remains a matter of controversy, whether autophagy promotes or prevents apoptosis after acute kidney injury (AKI) or stress [1, 6]. This probably depends on experimental conditions and on the cellular context. As a future perspective, autophagy may become an attractive target in the development of new renoprotective therapies. In this review, we will discuss the current evidence for autophagy involvement in various renal diseases. To ensure a good understanding of the molecular background, a general overview of the different autophagy mechanisms is presented.
Macroautophagy, chaperone-mediated autophagy, and microautophagy: an overview
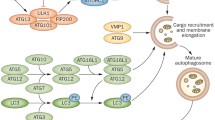
Three pathways of autophagy have been described: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). This differentiation is mainly based on their regulatory mechanisms (Fig. 1).

General overview of the three pathways of autophagy with their regulatory proceedings: chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy. CMA involves chaperone-dependent selection of soluble cytosolic proteins (in dark red) for degradation. The cytosolic chaperone heat shock cognate protein of 70 kDa (hsc70, in blue) recognises KFERQ motifs in substrate proteins, leading to targeting of the protein to the lysosomal surface. There, the complex binds to lysosome-associated membrane protein type 2A (LAMP-2A, in grey), leading to unfolding of the substrate protein and multimerisation of LAMP-2A. The latter forms an active translocation complex through which the substrate protein enters the lysosome, where lysosomal protease-based degradation takes place. Microautophagy involves engulfment of cytoplasmic cargo (in blue) by direct invagination of the lysosomal membranes into autophagic tubes. The macroautophagic pathway proceeds through several phases: initiation (formation of the phagophore from the omegasome); vesicle elongation forming autophagosome; autophagosome maturation; autophagosome-endosome fusion forming amphisome; amphisome-lysosome fusion forming autolysosome. In the latter, the entrapped cytosolic proteins (in green) and organelles such as mitochondria (in grey) are degraded. The phosphatidylinositol 3-kinase (PtdIns3K), Vps34, and its regulatory protein kinase Vps15, phosphorylate phosphatidylinositol to generate phosphatidylinositol 3-phosphate (PtdIns3P or PI3P). This results in PI3P-enriched omegasomes. Beclin 1 is a regulatory platform protein for this complex. Another initiating complex is the Unc-51-like kinase 1 and 2 (ULK1/2) complex, the mammalian homolog of yeast Atg1. Both FIP200 and Atg13 are critical for the correct localisation and stability of ULK1, and can enhance ULK1 kinase activity. AMP-activated protein kinase (AMPK) phosphorylates ULK1, and this modification is required for ULK1 activation after glucose deprivation. On the other hand, Atg13 and ULK1 are negatively regulated by the mTOR pathway via phosphorylation under nutrient-rich conditions. Autophagosomal elongation requires the microtubule-associated protein 1 light chain 3 (LC3/Atg8) conjugation system. The latter involves the conversion of LC3-I into phosphatidylethanolamine-conjugated LC3-II. AMP-activated protein kinase (AMPK) activates macro-autophagy as an energy sensor via mTOR inhibition and activation of ULK1.  = inhibition
= inhibition  = stimulation
= stimulation
Macroautophagy
Macroautophagy is the best-studied autophagic pathway, forming a degradation system for long-lived cytoplasmic proteins and dysfunctional organelles. Macroautophagy levels decline with age, leading to cellular age-related waste accumulation as a cause of the progressive ageing process [6]. The process starts with the formation of a structure on the endoplasmic reticulum, called the omegasome, that expands into a phagophore or isolation membrane, the precursor of the autophagosome. During phagophore elongation, cytoplasmic components are engulfed within this structure. Next, it closes to form a double-membrane vesicle, called an autophagosome. Autophagosomes first fuse with endosomes to form amphisomes. In turn, amphisomes will fuse with acidic lysosomes, forming autolysosomes, where the entrapped cytosolic contents are degraded [4].
Here, we summarise the main components of the autophagic machinery (Fig. 1). Several autophagy-related gene (Atg) proteins and vacuolar protein sorting gene (Vps) products are important for the initial sequestration process. Essential for omegasome formation is the Vps34-Vps15-Beclin 1 complex. The phosphatidylinositol 3-kinase (PtdIns3K), or Vps34, phosphorylates phosphatidylinositol to generate phosphatidylinositol 3-phosphate (PtdIns3P or PI3P) [7]. This results in PI3P-enriched omegasomes, acting as a cradle for the recruitment of autophagic proteins, and the formation of the phagophore [7, 8]. The mammalian homolog of yeast Atg6, Beclin 1, acts as a regulatory platform protein for the PtdIns3K complex by recruiting functional modulating proteins, including the anti-apoptotic Bcl-2 family of proteins [9, 10]. It is of note that there are two classes of Bcl-2 proteins: anti-apoptotic proteins (including Bcl-2, Bcl-XL, Bcl-w and Mcl-1) and pro-apoptotic BH3-only proteins (including BNIP3, Bad, Bik, Noxa, Puma, and BimEL) [11].
Another initiating complex, is the Unc-51-like kinase 1 and 2 (ULK1/2) complex, the mammalian homolog of yeast Atg1. The phosphorylation status of this complex plays a key role in the initial stages of autophagy, though the exact molecular mechanism is unknown. Both FIP200 and Atg13 are critical for the correct localisation and stability of ULK1, and can enhance ULK1 kinase activity. Furthermore, AMP-activated protein kinase (AMPK) directly phosphorylates ULK1 on several sites and this modification is required for ULK1 activation after glucose deprivation. On the other hand, Atg13 and ULK1 are negatively regulated by the mTOR pathway via phosphorylation under nutrient-rich conditions [12, 13].
Autophagosomal elongation requires the Atg5-Atg12 and the microtubule-associated protein 1 light chain 3 (LC3/Atg8) conjugation system. The latter involves the conversion of LC3-I into phosphatidylethanolamine-conjugated LC3-II: upon induction of autophagy, LC3-I is lipidated to form LC3-II, which is then incorporated into the autophagosomal membrane [14]. This conversion is indicative of autophagosome formation and is often used as a biochemical marker for evaluating autophagy [2]. It is important to highlight that an increase of LC3-II can signify stimulated autophagy but also autophagosome accumulation due to autophagy impairment. As the activity of autophagy is controlled by autophagosomal formation and autophagosomal degradation, the rate of the autophagosomal turnover is defined as “autophagic flux”. This is the true indicator of autophagic activity, for which LC3-I and LC3-II levels alone are not sufficient. Blocking either the synthesis or degradation of LC3-II using autophagic inhibitors distinguishes between increased autophagosomal formation and impaired autophagosomal degradation. For instance, wortmannin inhibits autophagosome formation and bafilomycin A1 stops LC3-II degradation.
The serine/threonine kinase AMPK activates macroautophagy as an energy sensor via mTOR inhibition and activation of ULK1 [11]. An alternative mTOR-independent macroautophagy regulator is the transcription factor EB (TFEB) [15]. TFEB is a controller of lysosomal biogenesis positively regulating genes belonging to the Coordinated Lysosomal Expression and Regulation (CLEAR) network. It was shown to activate a transcriptional program regulating major steps of the autophagic pathway with a net result of enhanced autophagic flux. In normal conditions, TFEB is localised to the cytoplasm. Nutrient starvation induces its nuclear translocation by inhibition of the extracellular signal-regulated kinase 2 (ERK2), belonging to the mitogen-activated protein kinases (MAPK). Genes known to play a role in autophagosome formation, autophagosome-lysosome fusion, and substrate degradation appear to be the direct targets of TFEB [15].
Chaperone-mediated autophagy
Chaperone-mediated autophagy (CMA) involves chaperone-dependent selection of soluble cytosolic proteins for degradation. The cytosolic chaperone heat shock cognate protein of 70 kDa (Hsc70) recognises CMA-targeting motifs in substrate proteins. This is a pentapeptide motif with a consensus sequence similar to KFERQ. Recognition by Hsc70 leads to targeting of the protein to the lysosomal surface. There, the complex binds to lysosome-associated membrane protein type 2A (LAMP-2A), leading to unfolding of the substrate protein and multimerisation of LAMP-2A. The latter forms an active translocation complex through which the substrate protein enters the lysosome, where lysosomal protease-based degradation takes place [16]. The unique features of this type of autophagy are the selectivity on the degraded proteins and the direct translocation of substrate proteins across the lysosomal membrane without the requirement for the formation of additional vesicles. The rate-limiting factor of this process is the LAMP-2A level. This is determined by its degradation by two proteases, a metalloprotease that cleaves the protein inside the lysosomal membrane and cathepsin A, which cleaves the intralysosomal portion of the protein. To a lesser extent, LAMP-2A levels are also determined by synthesis [17]. Ageing leads to less stable LAMP-2A, resulting in lower levels of CMA activity [16]. Primary CMA defects are described in lysosomal storage disorders such as cystinosis [6] and in neurodegenerative diseases such as Parkinson’s disease [18] and tauopathies [19]. In cancer cells, CMA has been shown to be upregulated [20]. There is a functional reciprocal crosstalk between CMA and macroautophagy, as CMA is most active in cells in which macroautophagy is least active [17]. Inhibiting either one of these processes leads to upregulation of the other, thus providing a constant baseline autophagy level [6].
Microautophagy
Microautophagy is a non-selective degradative process. It involves engulfment of cytoplasmic cargo by direct invagination of the lysosomal membranes into autophagic tubes. The maintenance of organelle size, membrane homeostasis, and cell survival under nitrogen restriction are the main functions of microautophagy. However, the exact physiological functions of microautophagy in mammalian cells are not completely understood [21].
Apoptosis and autophagy-dependent cell death
Historically, three types of cell death have been identified based on morphological criteria and regulating molecules, namely type I (apoptosis), type II (autophagic cell death), and type III (necrosis) [22]. We will focus on the first two types. Apoptosis is a process that executes degradation of whole cells instead of cellular components, as occurs upon degradation by autophagy. It is a vital factor for normal cell turnover, the immune system, embryonic development, and chemical-induced cell death. Inappropriate apoptosis, either too little or too much, is observed in various human pathologies. However, different cell types will have different responses to the same stimulus [23]. An apoptotic cell undergoes typical morphological changes, as described in Table 1. Apoptotic bodies, released during the budding process, consist of cytoplasm with tightly packed organelles with or without a nuclear fragment. These bodies are subsequently phagocytised by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes. The rapid clearance of apoptotic cells ensures minimal risk of detrimental inflammation [24]. There are two main apoptotic pathways: the extrinsic pathway and the intrinsic pathway. The extrinsic signalling pathway involves transmembrane receptor-mediated interactions. Here, tumor necrosis factor (TNF) receptor gene superfamily members have an important role, as they share similar cysteine-rich extracellular domains and have a cytoplasmic domain of approximately 80 amino acids called the “death domain.” The latter is critical in transmitting the death signal from the cell surface to intracellular signalling pathways [23]. The intrinsic signalling pathways are mitochondrial-initiated events, and involve diverse non-receptor-mediated stimuli that produce intracellular signals acting directly on targets within the cell, particularly the Bcl-2 family proteins. An additional pathway involves T-cell-mediated cytotoxicity and perforin-granzyme-dependent killing of the cell. The extrinsic, intrinsic, and granzyme B pathways converge on the same execution pathway, which is initiated by the cleavage of caspase-3.
In general, autophagy inhibits apoptosis as a protection mechanism against stressors before cellular disassembly. However, in case of autophagy deficiency or in response to intense and/or prolonged stress, there is a functional switch to pro-death autophagy-dependent mechanisms [3]. Moreover, there is a functional interplay between autophagy and apoptosis, enabling them to regulate each other’s activity both negatively and positively (Table 1). For instance, the interaction of Bcl-2 with Beclin1 prevents the association of the latter with the class III PtdIns 3-kinase, thereby inhibiting macroautophagy initiation. This can be reversed by agents mimicking the BH3 domain of Bcl-2. Similarly, activated apoptosis-related caspases can cleave autophagy-related proteins, such as Atg proteins and Beclin1, inhibiting autophagy [25]. In contrast, Atg12 conjugation to Atg3 enhances Bcl-XL expression, thereby inhibiting apoptosis. Unconjugated Atg12 can induce apoptosis by inhibiting Bcl-2 [26].
Table 1 gives an overview of the morphologic features, the key regulating and interacting molecules of apoptosis and autophagy.
Signalling pathways regulating autophagy
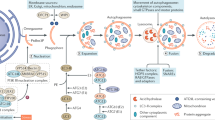
Several signalling pathways regulate autophagy in mammalian cells. An important player is mTOR, evolutionarily conserved from yeast to mammals, and negatively regulating autophagy [4, 5]. It exists in two functional multiprotein complexes: the rapamycin-sensitive mTOR complex 1 (mTORC1) and the rapamycin-insensitive mTOR complex 2 (mTORC2) (Fig. 2). However, prolonged rapamycin application can inhibit both [27]. The proteins forming both complexes, are described in Fig. 2. mTORC1 regulates cell growth through stimulation of protein anabolism, nucleotide biosynthesis, lipogenesis, glycolysis, and through inhibition of autophagy. This pathway is hyperactivated in a broad spectrum of disorders, including human cancers, metabolic diseases, and polycystic kidney disease [28, 29]. Less is known regarding mTORC2 function. It is likely to control cell growth by regulating lipogenesis, glucose metabolism, the actin cytoskeleton, and apoptosis [5, 30]. Growth factors stimulate both mTORC1 and 2, the first is also inhibited by stress and stimulated by nutrients. Energy status is translated to mTOR by AMPK, acting as a sensor of the cellular energy status [31]. AMPK is activated by the serine/threonine kinase LKB1, and this activation is called the LKB1-AMPK cascade [11]. mTORC1’s most proximal and direct activator is the small guanosine triphosphatase (GTPase) Ras homolog enriched in brain (Rheb). Rheb is a farnesylated GTPase anchored to the lysosomal surface and is inhibited by the tuberous sclerosis complex (TSC), which itself is influenced by Wnt signalling, hypoxia, inflammation, low energy status, and DNA damage [28, 31]. Growth-related kinases, such as Akt and p90 ribosomal protein S6 kinase, inhibit TSC function, thereby activating the Rheb–mTORC1 pathway. AMPK activates TSC function, inhibiting Rheb–mTORC1 activity and inducing macroautophagy. Consistently, in TSC-deficient cells, mTORC1 is in an active state and insensitive to inhibitory effects of growth factor deprivation [5]. Parallel but dominant to this Rheb-mediated pathway is the amino acid-induced activation of mTORC1. The latter is mediated via the RAS-related GTP-binding protein (Rag) proteins, heterodimeric GTPases consisting of either RagA or B, with either RagC or D. Amino acids convert heterodimers to the active heterodimeric conformation in which RagA or RagB is loaded with GTP and RagC or RagD is loaded with GDP. Rag proteins are regulated by lysosome-associated molecular machinery, monitoring amino acid availability and containing the multi-subunit Ragulator and the vacuolar ATPase [32]. The Ragulator complex has been implicated in binding the Rag GTPase heterodimer to the lipid bilayer [33]. It was recently shown that the lysosomal membrane protein human member 9 of the solute carrier family 38 (SLC38A9), a physical and functional component of the amino acid sensing Ragulator-Rag GTPase complex, positively regulates the mTORC1 pathway [32, 34]. Additionally, the proton-assisted amino acid transporter 1 (PAT1) or SLC36A1 has been shown to be an essential modulator of amino acid-dependent mTORC1 activation [33]. Activation via this pathway leads to the translocation of mTORC1 to the lysosomal surface where it can be further activated by Rheb, stimulating the mTORC1 kinase activity [5]. The regulation of autophagy by mTOR occurs via its substrates ULK1 and Atg13, both subunits of the autophagy-initiating ULK1 complex. By phosphorylating (and thereby inactivating) ULK1 and Atg13, mTORC1 inhibits the autophagy-initiation step [5, 31].

General overview of mTOR activators, inhibitors, and substrates. mTOR exists as two functional multiprotein complexes: the rapamycin-sensitive mTOR complex 1 (mTORC1) and the rapamycin-insensitive mTOR complex 2 (mTORC2). mTORC1 contains (a) the mTOR catalytic subunit, in which the drug-receptor complex FKBP12-rapamycin binding (FRB) domain is located, which is the interaction site for the intracellular rapamycin receptor FKBP12, (b) a substrate-recognising regulatory-associated protein of mTOR (RAPTOR), which recruits the mTORC1 substrates including S6 kinases, eukaryotic initiation factor 4E binding protein 1 (4EBP1) and Unc-51-like kinase 1 (ULK1), (c) G protein β-subunit-like protein (GβL), also known as mammalian lethal with SEC13 protein 8 (mLST8) and required for the nutrient- and rapamycin-dependent interaction between Raptor and mTOR, (d) the negatively regulating DEP domain-containing protein 6 (DEPDC6) or DEPTOR and (e) proline-rich Akt substrate of 40 kDa (PRAS40). mTORC2 consists of (a) mTOR, (b) the rapamycin-sensitive companion of mTOR (RICTOR), (c) stress-activated map kinase-interacting protein1 (SIN1), (d) protein observed with rictor (PROTOR), (e) G protein β-subunit-like protein, and (f) DEPDC6. mTORC1 regulates cell growth through stimulation of protein anabolism via inhibition of 4EBP1, ribosome biosynthesis via S6 kinase, and through inhibition of autophagy by inhibiting ULK1. Energy status is translated to mTOR by AMP-activated protein kinase (AMPK), which is itself activated by LKB1. mTORC1’s most proximal and direct activator is the Ras homolog enriched in brain (Rheb), which is inhibited by the tuberous sclerosis complex (TSC). TSC is influenced by AMPK, Wnt signalling, and growth-related kinases, such as Akt. Dominant to this Rheb-mediated pathway is the amino acid-induced activation of mTORC1, mediated via the RAS-related GTP-binding protein (Rag) proteins.  = inhibition
= inhibition  = stimulation
= stimulation
Role of autophagy in renal epithelial cell homeostasis
Podocyte
Unlike tubular cells, which have a high turnover and regeneration capacity, podocytes are post-mitotic cells, and in the presumed absence of regeneration, a decreasing number of podocytes predicts the progression of renal diseases and ageing [27]. Differentiated post-mitotic podocytes display both higher basal autophagy activity and higher mTORC1 activity compared to other glomerular cells. Although this seems contradictory, it can be explained as a coordinated spatial regulation by means of a distinct cellular compartment, named TOR-autophagy spatial coupling compartment or TASCC. TASCC is located in close proximity to the trans-Golgi network and provides high levels of lysosomes, autolysosomes, and mTOR. This spatial separation allows autophagosome formation to remain unaffected by mTORC1-dependent autophagy suppression [35]. This is a feature by which these cells can generate sufficient secretory proteins under a constant energy supply [5, 6]. The higher levels of basal autophagy compared to other glomerular cell types are considered as a cytoprotective quality-control machinery, on which podocytes are dependent to prevent cellular degeneration. As podocytes are post-mitotic cells, they cannot dilute possibly toxic cellular components into their daughter cells. They are in need of high basal autophagy levels for the removal of these components [6]. This is illustrated by a recent study in which mice harboring Atg7-deficient podocytes underwent a unilateral nephrectomy as a surgical model for acute nephron reduction. One day post-nephrectomy, these mice showed significantly higher levels of proteinuria, foot process effacement, and podocyte loss compared to wild-type mice [36]. Autophagy dysregulation in podocytes also has a clinical relevance in several lysosomal storage diseases, which are associated with the accumulation of storage products in podocytes, such as the X-linked Fabry disease. This disease is caused by deficient lysosomal hydrolase α-galactosidase A resulting in renal dysfunction, systemic vasculopathy, and cardiomyopathy. Fabry disease is shown to be associated with deregulated autophagy pathways, more specifically with impaired autophagosome maturation and thus compromised autolysosomal degradative capability [6, 37]. mTOR seems the key regulator of podocyte size control, particularly during development. It has been shown that loss of mTORC1 hampers normal differentiation of podocytes, displayed as partially effaced secondary foot processes without redistribution of slit diaphragm proteins [6]. Moreover, podocyte injury reactivates Notch, Wnt, and mTOR pathways. It is presumed that transient mTORC1 activation compensates for podocyte loss by hypertrophy of the remaining cells, while persistent mTORC1 activation leads to deregulated hypertrophy, which itself gives rise to podocyte loss and glomerulosclerosis [27]. This double-edged role of mTORC1 in glomerular function leaves rapamycin both a promising medicine and a toxin. On the other hand, mTORC2-Akt2 activation plays a critical role in podocyte survival. mTORC2-Akt2 activation could be observed in biopsy tissue from kidney transplant patients and rapamycin was shown to prevent the activation of mTORC2-Akt2 in these patients. The latter was associated with increased glomerular apoptosis. This could be a contributing factor to rapamycin-induced proteinuria, in concert with mTORC1 [27]. It would be interesting to compare the use of specific mTORC1 inhibitors to mTOR inhibitors.
Tubular cells
In contrast to podocytes, basal autophagy activity in tubular cells, which are non-post-mitotic, is presumably low under normal physiological conditions [38]. In the mature tubule, CMA is the predominant form of autophagy in the basal state [17]. Autophagy induction in tubular cells, especially in proximal tubular cells, plays an important role in protecting cells from stress as proteinuria-induced apoptosis and ischemic AKI, as shown in both in vitro and in vivo experimental models [5]. How exactly autophagy executes its protective role in renal tubular cells undergoing injury or apoptosis remains unclear. Oxidative stress and endoplasmic reticulum stress through the unfolded protein response and intracellular Ca2+ have been implicated in autophagy regulation of renal tubular cells [39]. The observation that autophagy occurs prior to apoptosis in these cells during AKI suggests that autophagy is an early response of the cells to stress and not a result of apoptosis. An in vivo study has suggested mTOR pathway involvement in tubular repair after AKI. In this study, rapamycin compromised recovery by inducing apoptosis and inhibiting proliferation of tubular cells [39]. Elevated mTORC1 levels were also seen in another study in the model of kidney ischemia-reperfusion (IR), together with decreased AMPK activity. However, agonist induction of AMPK activity with AICAR or metformin prior to ischemia increased LC3-I, normalised mTORC1 activity, and significantly attenuated apoptotic marker expressions. The increase of cytosolic LC3-I was not translated into a marked LC3-II response until the IR stimulus, implying that agonists prime the system to allow for a rapid increase in autophagy that only occurs in response to appropriate stimulation [14]. These data suggest that physiologically occurring autophagy levels after renal IR injury are insufficient to counter the cellular insults in IR, and therefore may contribute to cell death during AKI [14].
Cilia
Recently, there has been increasing evidence for a relationship between autophagy and the formation of primary cilia. The latter are the solitary, microtubule-based, non-motile structures growing from the centriole and protruding from the plasma membrane of many cell types, sensing and transducing input from the extracellular environment (mechanical stress, nutrients, growth factors, and Ca2+ changes) into different cellular signalling pathways [40]. The fact that nutrient deprivation is a primary stimulus shared by both autophagy and primary cilia formation supports the proposed relationship between the two processes [41]. Intraflagellar transport (IFT) proteins that are required for ciliary elongation and function have been shown to be involved in autophagosome biogenesis in response to cilium-dependent signalling [42]. Cell lines in which IFT proteins (IFT20 or IFT88) were knocked out became defective in both ciliogenesis and in autophagosome formation, illustrating that functioning IFT proteins are necessary for both processes. It was further demonstrated that signalling via the Hedgehog pathway is required for cilia-induced autophagy [42]. Moreover, it was observed that components of the autophagic machinery are located at the primary cilium axoneme and basal body [42].
Besides primary cilia-induced autophagosome formation, it was found that autophagy regulates biogenesis of cilia by degrading certain proteins involved in cilia formation. This includes both components essential for ciliogenesis, such as IFT20, and negative ciliogenesis regulators, such as oral facial digital syndrome 1 (OFD1), depending upon experimental conditions [40]. Atg5-null cells, which are defective in autophagy, formed cilia at a faster rate and of longer length than wild-type cells [42]. As the inhibition of autophagy correlated with increased IFT20 levels, basal autophagy seems to negatively regulate ciliogenesis by degrading IFT20. Starvation-induced autophagy, however, promoted ciliogenesis by degrading a particular pool of OFD1, a protein localised at the centrioles and the centriolar satellites, interacting with LC3 [43]. In autophagy-deficient mouse embryonic fibroblasts (Atg5 or Atg3-null MEFs), OFD1 accumulated at the centriolar satellites, leading to fewer and shorter primary cilia. It was suggested that removal of OFD1 by autophagy may represent a general mechanism to promote ciliogenesis in mammalian cells [43]. Thus, basal autophagy and autophagy induced by signalling from the cilia may negatively regulate cilia growth through degradation of IFT proteins [42], while starvation-induced autophagy promotes ciliogenesis by specific degradation of the OFD pool at the centriolar satellites [43]. This switch highlights the significance of the interplay between autophagy and ciliogenesis for cellular homeostasis (Fig. 3). These findings show that autophagy must be considered as a factor in the development of conditions associated with defective cilia, called ciliopathies, such as autosomal dominant polycystic kidney disease (ADPKD) [6, 41]. Mutations in various ciliary proteins, including IFT88 and IFT20, can result in cyst formation [40]. ADPKD is characterised by a disturbance in many signalling pathways, several of which are directly related to the regulation of autophagy, e.g., mTOR signalling and intracellular Ca2+ signalling [44–46]. In agreement with a specific role for Ca2+ signalling, it was recently demonstrated that primary cilia are specialised Ca2+ signalling organelles [47] and the molecular identity of a Ca2+ channel in primary cilia has been described [48].

The interplay between autophagy and ciliogenesis. Nutrient deprivation is a primary stimulus shared by both autophagy and primary cilia formation [41]. Moreover, intraflagellar transport (IFT) proteins, which are required for ciliary elongation and function, are involved in autophagosome biogenesis in response to cilium-dependent signalling [42]. Besides primary cilia-induced autophagosome formation, autophagy regulates biogenesis of cilia by degrading certain proteins involved in cilia formation. Basal autophagy and autophagy induced by signalling from the cilia negatively regulate ciliogenesis by degrading IFT20, an essential component for ciliogenesis. Starvation-induced autophagy, however, promotes ciliogenesis by degrading a particular pool of the negative ciliogenesis regulator oral facial digital syndrome 1 (OFD1) [43]. This switch highlights the significance of the interplay between autophagy and ciliogenesis for cellular homeostasis.  = inhibition
= inhibition  = stimulation
= stimulation
Role of autophagy in renal disease
In this part, we will give an overview of the evidence for autophagy involvement in various renal diseases, as shown in Table 2.
Glomerulosclerosis
As stated earlier, autophagy plays a cytoprotective role in controlling the progression of podocytopathies. This is illustrated by a study comparing renal biopsies from patients with minimal change disease, in which there are no alternations in podocyte number and which has a benign prognosis; versus patients with focal segmental glomerulosclerosis (FSGS), which leads to podocyte loss and a poorer overall prognosis. Glomeruli, and more specifically the podocytes, from minimal change disease patients showed higher levels of Beclin 1-mediated autophagic activity than those from FSGS patients. Repeat renal biopsies in minimal change disease patients showed that those maintaining these high autophagy levels retained minimal change status, while those patients with decreasing levels of autophagy progressed to FSGS [49]. Moreover, in vitro and in vivo studies showed that inhibition of autophagy by treatment with 3-methyladenine or chloroquine enhanced puromycin aminonucleoside-induced podocyte apoptosis, while autophagy promotion via rapamycin administration decreased apoptosis [49]. Another recent study showed that mice with loss-of-function mutations in ATG5 or ATG7, developed histologic and clinical characteristics of human FSGS during nephrogenesis. In these animals, mitochondrial dysfunction and endoplasmic reticulum stress was found both in podocytes and tubular epithelial cells [50]. Another study showed that podocyte-specific conditional ATG5 knockout mice developed albuminuria and late-onset glomerulosclerosis [35]. These reports presume that promoting or maintaining podocyte autophagic activity during early disease stages is a potential therapeutic strategy for delaying the progression of podocytopathies.
Renal interstitial fibrosis
In progressive kidney diseases, fibrosis represents the common pathway to end-stage renal disease (ESRD), regardless of the initial causes. It has been shown in both in vitro and in vivo studies that the upregulation of transforming growth factor (TGF)-β plays a central role in the pathogenesis of renal fibrosis, with TGF-β1 as its most abundant isoform. Potent sources of this protein are either macrophages or injured tubular epithelium. Via downstream signalling pathways, TGF-β1 induces type I collagen and fibronectin production in interstitial fibroblasts [51]. The balance between synthesis and degradation of collagen is crucial for the maintenance of tissue homeostasis. Other important players in the development of fibrosis are the mesangial cells. In response to injury, they proliferate and produce excessive extracellular matrix. It has been shown that TGF-β1 induces autophagy in mesangial cells, thereby negatively regulating matrix production by the degradation of intracellular type I collagen [51]. Thus, TGF-β1 functions both as an inducer of collagen synthesis and as an inducer of autophagy and subsequent collagen degradation. Even more intriguingly, TGF-β can also activate the mTOR pathway via PI3K/Akt, and therefore may exert both stimulatory and inhibitory effects on autophagy, probably depending on the specific cell type and context [51]. From in vivo studies [51], it seems that both upregulation and downregulation of autophagy may be involved in fibrogenesis. For instance, AMPK induction by metformin has been shown to be a promising therapeutic target in reducing renal fibrosis [52].
Diabetic nephropathy
Diabetes type 1 and 2 have been shown to correlate with increased mTORC1 activity, and rapamycin prevents the development of diabetic nephropathy (DN) as glomerular hypertrophy, mesangial expansion, glomerular basement membrane thickening, and renal macrophage recruitment were observed less in diabetic animal models treated with rapamycin [27, 35]. Moreover, mTORC1 upregulation was found in podocytes of both animal models and humans with DN [6]. These findings suggest that mTORC1 hyperactivation is crucial for the onset or progression of DN. Therefore, mTORC1 may be a therapeutic target in patients with DN, although it is currently unacceptable because of the previously described adverse effects. Furthermore, obesity-mediated autophagy deficiency could be one of the elements explaining the vulnerability of proximal tubular cells in type 2 diabetic kidneys. Moreover, obesity-mediated autophagy is highly associated with mTORC1 hyperactivation [53]. This is illustrated by different animal and human studies. Autophagy activity was significantly suppressed in the kidneys of streptozotocin-induced diabetic mice, high-fat-diet-induced obese mice, and Wistar fatty rats, leading to the accumulation of damaged molecules and organelles, including p62 protein and damaged mitochondria. The protein p62 or sequestosome 1 is a selective substrate of macroautophagy that links ubiquitinated proteins to LC3. In case of inhibited macroautophagy, p62 accumulates in cells [11]. These findings were confirmed in renal biopsy specimens from patients with obesity and/or type 2 diabetes mellitus. Therefore, a calorie-restricted regimen, activating autophagy, should become a potent therapeutic strategy to prevent DN, as it has been shown to improve renal damage in Wistar fatty rats [53]. Another autophagy inducer and mTORC1 interactor likely to be suppressed in the development of DN is the energy-sensing kinase AMPK. This suggests that AMPK activation by agents such as metformin and resveratrol may be a target for restoring autophagy activity, even in diabetic kidneys [53].
Transplantation/ischemia
Renal ischemia is a major cause of AKI and inevitable in renal transplantation. This will coerce the tissue into anaerobic metabolism for survival, but paradoxically this sets the stage for oxidative damage upon blood reflow, triggering inflammation and cell death. This process, known as IR injury, is a major hurdle during kidney transplantation and significantly influences short- and long-term graft function and rejection. In the kidney, the proximal tubular cells are most susceptible to IR stress. Considering the role of autophagy in the proximal tubular cell balance between life and death during starvation, and the occurrence of hypoxia (ischemia) and oxidative stress (reperfusion), its influence on the transplanted kidney is undeniable [54]. However, the exact outcome of autophagy stimulation in renal IR injury is still a matter of debate. Although it may seem logical to reinforce autophagy during renal IR as a protective mechanism to maintain energy production during the hypoxic starvation period of ischemia and/or counteract oxidative damaged proteins and organelles during reperfusion [55–58], several reports have made opposite observations in which autophagy contributed to renal IR damage [59–64]. Possible reasons for these conflicting findings are likely multiple. First, the role of autophagy is deduced from the effect of chemical autophagy stimulation (rapamycin [62]) or suppression (3-methyladenine, chloroquine [58, 61]) in experimental animal models of renal IR injury. These compounds, however, are not specific and affect other processes as well. For instance, independent of autophagy stimulation, mTOR inhibition by rapamycin or everolimus will suppress protein transcription and translation as well as the cell cycle. These effects may account for the damaging effects observed during renal IR [62]. Similarly, autophagy suppression through class III PtdIns3K inhibition by 3-methyladenine will also affect the class I PtdIns3K /Akt/mTOR pathway and suppress the cell cycle. Notably, 3-methyladenine had both protective [61] and detrimental effects [58] during renal IR. These differences can possibly be explained by the different experimental models used: besides the obvious differences based on species, differences are also observed depending on gender and age. Females are often considered to be more resistant to ischemic damage compared to males [65, 66] and autophagy in the proximal tubular cells is clearly regulated by sex hormones [67]. Moreover, autophagy dysfunction is a recurring feature upon ageing [68]. As such, autophagy dysfunction could also partially account for the higher degree of early immune responses after kidney transplantation of renal grafts of older rat donors [69]. The generation of proximal tubule-specific Atg5- and Atg7-knockout mice has largely overcome the problem of the non-specificity of the chemical autophagy modulators. Using these more specific models, autophagy was considered as a protective mechanism during renal IR [55–57]. However, it should be noted that these knockout mice were subjected to relatively mild ischemic stress, that is 25 or 40 min of ischemia time. This is important since a striking association is observed in the experimental renal IR models between the length of ischemia and the proposed role of autophagy [70]. Autophagy was considered protective when ischemic duration was short (20–40 min), but detrimental when ischemic stress was severe (40–60 min). As such, the outcome of autophagy modulation may depend on the length of ischemia in the animal model. Longer ischemia duration could sensitise kidney cells to autophagy-dependent cell death or autophagy impairment associated with autophagosomal accumulation post-reperfusion. This may lead to increased injury upon extra autophagy stimulation or reduced injury by autophagy suppression [70]. As such, more information is needed regarding the exact role of autophagy upon varying degrees of IR stress before the modulation of autophagy can be considered as a therapeutic strategy to attenuate renal IR injury. Besides the protective versus detrimental role of autophagy in renal IR injury, autophagy modulation will also have important effects on the rejection or tolerance of the kidney graft due to its role in immunity [71]. The latter is dependent on the cell type and the immunological stimulus. The outcome of systemic autophagy stimulation on rejection or tolerance is difficult to predict. Local autophagy modulation may be a preferred strategy to alleviate late graft rejection after transplantation.
Cystic diseases
ADPKD is caused by mutations in two genes: PKD1 or PKD2, encoding the polycystin-1 (PC1) and polycystin-2 (PC2) ciliary proteins, respectively. These proteins are involved in cellular repair and growth mechanisms. Epithelial cyst-lining cells expressing mutated PC1 display higher rates of cell proliferation [40]. However, their exact role is complex and not fully clarified [40]. Extensive study of the mechanism of cyst development and growth, in vitro and in vivo, has shown that multiple molecular parameters and signalling pathways are involved, such as mTOR, vasopressin, cyclic adenosine monophosphate, several growth factors, caspases, and apoptosis [41]. Many of these are also involved in autophagy and apoptosis regulation. Moreover, various cyst-inhibiting compounds, such as mTOR inhibitors, metformin [6], triptolide [72], and curcumin [41] are autophagy inducers as well. This has led to the hypothesis that suppression of autophagy may be associated with increased apoptosis, and might play a role in cyst formation and growth [40, 41]. First, the mTOR pathway, the main cellular suppressor of autophagy, is shown to be activated in ADPKD rodent models and in human cells. Mutated PC1 activates the MEK/ERK pathway, which causes phosphorylation of Tuberin on Serine 664. In addition, Rheb is no longer inhibited by the LKB1/AMPK axis because of ciliary dysfunction. Indeed, ADPKD is a ciliopathy. It has been shown that normal ciliary bending is required for downregulation of the mTOR pathway, independently of both Ca2+ influxes and Akt [41]. This is disturbed in ADPKD. Although rapamycin was shown to be an effective therapeutic agent in ADPKD rodent models, clinical trials in ADPKD patients so far have shown no functional benefit of rapamycin on renal function [27]. Second, localised hypoxia is present in ADPKD because of cyst expansion. This was demonstrated by higher levels of erythropoietin in ESRD ADPKD patients, and by higher levels of hypoxia-inducible factor-1α (HIF-1α) in renal cyst lining epithelial cells of different rodent disease models with varying severity of cyst formation [73]. Moreover, evidence is mounting that apoptotic cell death promotes cyst growth in polycystic kidney disease [41]. Next, in tubular cyst-lining cells, the presence of autophagosomes and LC3-II was observed [73]. However, treatment with the lysosomal degradation inhibitor bafilomycin A1 in congenital polycystic kidney mice did not have an effect on LC3-II levels, while in wild-type mice it resulted in a significant increase of LC3-II. Another study shows that mouse kidney cells expressing mutated PC1 failed to induce autophagy in response to glucose deprivation. Instead, these cells displayed high levels of apoptosis. Treating the cells with rapamycin led to a higher cell survival [74]. Taken together, these studies suggest an impairment of autophagy in ADPKD, resulting from a block of autophagosome-lysosome fusion and degradation [41, 73], confirming the earlier stated hypothesis that suppression of autophagic flux may be a feature of ADPKD. However, the mechanisms of crosstalk between autophagy induction, proliferation, and apoptosis in ADPKD still remain to be unraveled. Recently, it has been demonstrated that PC1 controls PC2 degradation in an autophagy-dependent way, and that the pathogenic PC1 mutants fail to induce this function [75]. Based on current knowledge, induction of autophagy may have a therapeutic role in decreasing cyst development [41].
Lysosomal storage disease: cystinosis
Nephropathic cystinosis is a rare autosomal recessive lysosomal storage disease caused by mutations in the CTNS gene that encodes a lysosomal cystine transporter, cystinosin [76]. Early clinical manifestations of cystinosis include the renal Fanconi syndrome, a generalised proximal tubular dysfunction [77]. Podocyte dysfunction is also present in cystinosis, leading to intermediate and high molecular weight proteinuria and pathological changes in the glomeruli, including podocyte foot process effacement and glomerular sclerosis [78]. Current treatment of cystinosis is based on the cystine-lowering drug cysteamine [76]. Cysteamine therapy allows patients to survive into adulthood, attenuating the disease’s progression, but does not cure Fanconi syndrome and the eventual development of kidney damage. Hence, it has been hypothesised that the disease pathogenesis is more complex than merely cystine accumulation in the lysosomes [79]. Several lines of evidence point to the possible role of autophagy and altered mTOR signalling in cystinosis. First indications of cystinosin implication in mTOR regulation came from studies of protein interactions performed on yeast. The yeast analogue of cystinosin, ERS1, interacts with the EGO (exit from growth arrest) complex residing on the vacuole, which is the yeast analogue of the lysosome, regulating the yeast TOR signalling [80, 81]. Recent studies provide more evidence for the autophagy dysfunction in cystinosis. It has been demonstrated that cystinosis fibroblasts and proximal tubular cells have increased autophagy of mitochondria, termed mitophagy [82, 83]. Cystinosis cells have increased amounts of autophagosome markers LC3-II and SQSTM1/p62, suggestive of disrupted autophagic flux [84]. Moreover, the expression of clusterin, a protein involved in nephropathic cystinosis, overlaps with the expression of apoptotic and autophagy proteins [85]. Detailed study of autophagy in a cystinosis mouse model has shown, however, that the autophagic flux and mTOR signalling were not compromised. However, a decreased expression and abnormal localisation of LAMP-2A, the lysosomal receptor responsible for CMA, was described. Correspondingly, CMA was impaired in cystinosin-deficient cells, as demonstrated by measurement of the degradation of GAPDH, one of the CMA substrates, and this impairment could not be corrected by cysteamine treatment [86].
The available data clearly demonstrate that cystinosis is associated with altered autophagy, and such association is not entirely dependent on the lysosomal storage of cystine. More studies are needed to unravel the exact mechanisms underlying the impaired autophagy and to explore its potential as a therapeutic target.
Therapeutic targets and clinical implications
Undoubtedly, autophagy is an attractive target for developing new renoprotective treatments, and also as a preventive renal anti-ageing mechanism [6]. There are, however, challenges that must be addressed before this strategy can be considered tenable and feasible. The known autophagy inhibitors, rapamycin, bafilomycin, chloroquine, and 3-methyladenine are not favorable compounds. As their biological effects cover more than just autophagy regulation, the net effect may paradoxically be an aggravation of tissue injuries [3]. More specific and selective regulators of the autophagic machinery could avoid this. Moreover, intermittent upregulation of autophagy may be more effective with fewer side effects than chronic use of such strategy. The AMPK inducer metformin could be a promising therapeutic agent, though further studies are necessary. However, modulating autophagy will remain an utterly challenging quest, as the borderline between renal cytoprotection and induction of apoptosis is a narrow one, which will depend on several complex factors such as timing, duration and intensity of autophagy induction [39]. Therapeutically, it will be very important to determine an optimal condition and therapeutic window in which induction of autophagy would yield protective effects. The technical issue remaining to be resolved before autophagy can enter the clinics as a parameter, is the ability to monitor autophagy in clinical practice for both diagnosis and therapeutic counselling, in the right tissue and with correct timing. Given that autophagy is a dynamic process, regulated within labile and short-lived networks of protein–protein interactions, it must be appreciated in terms of autophagic flux and requires chemical modulators to inhibit the degradation of autophagosomes by lysosomes. It is virtually impossible to characterise this flux using static biopsy specimens and, moreover, the complexity of this autophagic flux is incompatible with routine diagnostic tools.
Conclusions
Tremendous progress has been made in understanding the molecular mechanism and signalling pathways of autophagy from yeast to mammals. Defective autophagy has been shown to be associated with a broad variety of pathophysiological conditions. Renal autophagy research is in its infancy relative to disciplines such as oncology and neurology. Based on in vivo models of acute or chronic kidney injury, autophagy is regarded as a renoprotective factor in the development and progression of renal diseases and renal ageing. However, a defect in autophagy regulation itself is not (yet) proven to be able to cause human kidney diseases. Moreover, the role of autophagy in renal disease pathogenesis is likely multifaceted and complex: depending on experimental conditions, autophagy can be either protective or detrimental. The intricate interplay and cross regulation between autophagy and apoptosis pathways makes it difficult to predict how autophagy contributes to the life-and-death decisions of a stressed cell.
Future perspectives
Autophagy is an attractive target for developing new renoprotective treatments. Ideally, this should be achieved by more-specific and selective regulators of the autophagic machinery than the currently known compounds. Moreover, these should be used within an optimal therapeutic window to avoid adverse effects. For this, a validated tool to monitor autophagy in clinical practice is essential, which should incorporate a dynamic autophagic flux measurement, but this is not yet available for application in clinical conditions. Based on the current exciting new developments regarding the role of autophagy in kidney health and disease, we expect more advances to follow in the near future. These may lead to new therapeutic approaches in many renal diseases.
Key summary points
-
In cells, there are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy, which are distinguished mainly by their regulatory triggers.
-
In kidneys, autophagy plays important roles in the homeostasis and survival of renal cells such as podocytes and tubular epithelial cells.
-
Autophagy also plays an important role in acute and chronic kidney disease and renal ischemia.
-
The role of the regulation of autophagy in renal pathologies such as the podocytopathies, ciliopathies, and acute kidney injury is not yet completely understood.
-
Autophagy represents an interactive target for therapy but needs further research to delineate better its role.
Multiple choice questions (answers are provided following the reference list)
-
1.
Which of the following compounds does NOT induce autophagy:
-
a.
mTOR inhibitor
-
b.
Metformin
-
c.
Curcumin
-
d.
Chloroquine
-
e.
Caspase inhibitor
-
f.
Cyclin-dependant kinase inhibitors
-
g.
Triptolide
-
a.
-
2.
One of the following sentences is incorrect:
-
a.
Macroautophagy is a degradation system for long-lived cytoplasmic proteins and dysfunctional organelles
-
b.
Microautophagy is a selective degradative process. It involves engulfment of cytoplasmic cargo by direct invagination of the lysosomal membranes into autophagic tubes
-
c.
The unique features of chaperone-mediated autophagy are the selectivity on the degraded proteins and the direct translocation of substrate proteins across the lysosomal membrane without the requirement for the formation of additional vesicles
-
d.
Autophagy is a major protective mechanism allowing cell survival in response to multiple stressors and helping organisms to defend against degenerative, inflammatory, infectious, and neoplastic diseases
-
a.
-
3.
What is the significance of increased LC3-II in a sample?
-
a.
Autophagy stimulation
-
b.
Autophagy suppression
-
c.
Suppressed autophagic flux
-
d.
Enhanced autophagic flux
-
e.
Either autophagy stimulation or suppressed autophagic flux
-
f.
Either autophagy stimulation or enhanced autophagic flux
-
a.
-
4.
How do podocytes and tubular cells differ in autophagy dynamics?
-
a.
Podocytes are more dependent on CMA than tubular cells
-
b.
Podocytes show no sign of macroautophagy in normal physiological conditions
-
c.
Tubular cells are more dependent on CMA than podocytes
-
d.
Tubular cells show no signs of macroautophagy in starved conditions
-
a.
-
5.
How do autophagy and apoptosis interconnect? (2 answers are correct)
-
a.
Autophagy degrades the pro-apoptotic caspases
-
b.
Caspases cleave and inactivate different autophagy proteins
-
c.
Autophagy degrades damaged mitochondria, a source of apoptosis initiation via the intrinsic pathway
-
d.
Pro-apoptotic proteins Bcl-2 family proteins Bax and Bak bind and inhibit Beclin1, thereby preventing autophagy
-
a.
Abbreviations
- ADPKD:
-
Autosomal dominant polycystic kidney disease
- AKI:
-
Acute kidney injury
- AMPK:
-
AMP-activated protein kinase
- Atg:
-
Autophagy-related gene
- CMA:
-
Chaperone-mediated autophagy
- DN:
-
Diabetic nephropathy
- ESRD:
-
End-stage renal disease
- FSGS:
-
Focal segmental glomerulosclerosis
- GTPase:
-
Guanosine triphosphatase
- Hsc70:
-
Heat shock cognate protein of 70 kDa
- IFT:
-
Intraflagellar transport
- IPMK:
-
Inositol polyphosphate multikinase
- IR:
-
Ischemia-reperfusion
- LAMP-2A:
-
Lysosome-associated membrane protein type 2A
- LC3:
-
Microtubule-associated protein light chain 3
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
mTOR complex 1
- mTORC2:
-
mTOR complex 2
- OFD1:
-
Oral facial digital syndrome 1
- PAT1:
-
Proton-assisted amino acid transporter 1
- PC1/ PC2:
-
Polycystin-1 / polycystin-2
- Rag:
-
RAS-related GTP-binding protein
- RAPTOR:
-
Regulatory associated protein of mTOR
- Rheb:
-
Ras homolog enriched in brain
- TFEB:
-
Transcription factor EB
- TGF:
-
Transforming growth factor
- TSC:
-
Tuberous sclerosis complex
- ULK1:
-
Unc-51 like kinase 1
- Vps:
-
Vacuolar protein sorting gene
References
Dong Z (2014) Introduction: autophagy in kidneys. Semin Nephrol 34:1
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:651–662
Fougeray S, Pallet N (2015) Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol 11:34–45
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435
Inoki K (2014) mTOR signaling in autophagy regulation in the kidney. Semin Nephrol 34:2–8
Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, Ravichandran K, Susztak K, Yoshida S, Dong Z (2012) Emerging role of autophagy in kidney function, diseases and aging. Autophagy 8:1009–1031
Simonsen A, Stenmark H (2008) Self-eating from an ER-associated cup. J Cell Biol 182:621–622
Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6:506–522
Kang R, Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18:571–580
Decuypere JP, Parys JB, Bultynck G (2012) Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 1:284–312
Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, Debnath J, Deretic V, Elazar Z, Eskelinen EL, Finkbeiner S, Fueyo-Margareto J, Gewirtz D, Jaattela M, Kroemer G, Levine B, Melia TJ, Mizushima N, Rubinsztein DC, Simonsen A, Thorburn A, Thumm M, Tooze SA (2011) A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7:1273–1294
Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22:132–139
Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305
Decleves AE, Sharma K, Satriano J (2014) Beneficial effects of amp-activated protein kinase agonists in kidney ischemia-reperfusion: autophagy and cellular stress markers. Nephron Exp Nephrol 128:98–110
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22:407–417
Franch HA (2014) Chaperone-mediated autophagy in the kidney: the road more traveled. Semin Nephrol 34:72–83
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118:777–788
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18:4153–4170
Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM (2011) Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 3:109–117
Li WW, Li J, Bao JK (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69:1125–1136
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22:367–376
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Chan FK, Luz NF, Moriwaki K (2015) Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol 33:79–106
Li M, Tan J, Miao Y, Lei P, Zhang Q (2015) The dual role of autophagy under hypoxia-involvement of interaction between autophagy and apoptosis. Apoptosis 20:769–777
Tait SW, Ichim G, Green DR (2014) Die another way–non-apoptotic mechanisms of cell death. J Cell Sci 127:2135–2144
Grahammer F, Wanner N, Huber TB (2014) mTOR controls kidney epithelia in health and disease. Nephrol Dial Transplant 29(Suppl 1):i9–i18
Shaw RJ (2013) Cell biology. GATORs take a bite out of mTOR. Science 340:1056–1057
Mekahli D, Decuypere JP, Sammels E, Welkenhuyzen K, Schoeber J, Audrezet MP, Corvelyn A, Dechenes G, Ong AC, Wilmer MJ, van den Heuvel L, Bultynck G, Parys JB, Missiaen L, Levtchenko E, De Smedt H (2014) Polycystin-1 but not polycystin-2 deficiency causes upregulation of the mTOR pathway and can be synergistically targeted with rapamycin and metformin. Pflugers Arch 466:1591–1604
Betz C, Hall MN (2013) Where is mTOR and what is it doing there? J Cell Biol 203:563–574
Jewell JL, Guan KL (2013) Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 38:233–242
Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, Wang T, Bar-Peled L, Zoncu R, Straub C, Kim C, Park J, Sabatini BL, Sabatini DM (2015) Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347:188–194
Ogmundsdottir MH, Heublein S, Kazi S, Reynolds B, Visvalingam SM, Shaw MK, Goberdhan DC (2012) Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS One 7, e36616
Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, Scorzoni S, Filipek PA, Huber KV, Bigenzahn JW, Heinz LX, Kraft C, Bennett KL, Indiveri C, Huber LA, Superti-Furga G (2015) SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519:477–481
Hartleben B, Wanner N, Huber TB (2014) Autophagy in glomerular health and disease. Semin Nephrol 34:42–52
Oliva Trejo JA, Asanuma K, Kim EH, Takagi-Akiba M, Nonaka K, Hidaka T, Komatsu M, Tada N, Ueno T, Tomino Y (2014) Transient increase in proteinuria, poly-ubiquitylated proteins and ER stress markers in podocyte-specific autophagy-deficient mice following unilateral nephrectomy. Biochem Biophys Res Commun 446:1190–1196
Liebau MC, Braun F, Hopker K, Weitbrecht C, Bartels V, Muller RU, Brodesser S, Saleem MA, Benzing T, Schermer B, Cybulla M, Kurschat CE (2013) Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS One 8, e63506
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111
Livingston MJ, Dong Z (2014) Autophagy in acute kidney injury. Semin Nephrol 34:17–26
Orhon I, Dupont N, Pampliega O, Cuervo AM, Codogno P (2015) Autophagy and regulation of cilia function and assembly. Cell Death Differ 22:389–397
Ravichandran K, Edelstein CL (2014) Polycystic kidney disease: a case of suppressed autophagy? Semin Nephrol 34:27–33
Pampliega O, Orhon I, Patel B, Sridhar S, Diaz-Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM (2013) Functional interaction between autophagy and ciliogenesis. Nature 502:194–200
Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502:254–257
Mekahli D, Parys JB, Bultynck G, Missiaen L, De Smedt H (2013) Polycystins and cellular Ca2+ signaling. Cell Mol Life Sci 70:2697–2712
Decuypere JP, Kindt D, Luyten T, Welkenhuyzen K, Missiaen L, De Smedt H, Bultynck G, Parys JB (2013) mTOR-Controlled autophagy requires intracellular Ca(2+) signaling. PLoS One 8, e61020
Decuypere JP, Bultynck G, Parys JB (2011) A dual role for Ca(2+) in autophagy regulation. Cell Calcium 50:242–250
Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE (2013) Primary cilia are specialized calcium signalling organelles. Nature 504:311–314
DeCaen PG, Delling M, Vien TN, Clapham DE (2013) Direct recording and molecular identification of the calcium channel of primary cilia. Nature 504:315–318
Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong Y, Zhang C, Zen K, Liu Z (2014) Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol 234:203–213
Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, Shankland SJ, D’Agati VD, Duffield JS (2014) Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26:1040–1052
Ding Y, Choi ME (2014) Regulation of autophagy by TGF-beta: emerging role in kidney fibrosis. Semin Nephrol 34:62–71
Kim H, Moon SY, Kim JS, Baek CH, Kim M, Min JY, Lee SK (2015) Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am J Physiol Ren Physiol 308:226–236
Kume S, Yamahara K, Yasuda M, Maegawa H, Koya D (2014) Autophagy: emerging therapeutic target for diabetic nephropathy. Semin Nephrol 34:9–16
Pallet N (2014) Emerging roles of autophagy in the stressed kidney allograft. Semin Nephrol 34:34–41
Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, Huber TB (2012) Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 8:826–837
Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y (2011) Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22:902–913
Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z (2012) Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82:1271–1283
Jiang M, Liu K, Luo J, Dong Z (2010) Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 176:1181–1192
Yeh CH, Hsu SP, Yang CC, Chien CT, Wang NP (2010) Hypoxic preconditioning reinforces HIF-alpha-dependent HSP70 signaling to reduce ischemic renal failure-induced renal tubular apoptosis and autophagy. Life Sci 86:115–123
Wu HH, Hsiao TY, Chien CT, Lai MK (2009) Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J Biomed Sci 16:19
Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, Imai E (2008) Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 368:100–106
Nakagawa S, Nishihara K, Inui K, Masuda S (2012) Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur J Pharmacol 696:143–154
Isaka Y, Suzuki C, Abe T, Okumi M, Ichimaru N, Imamura R, Kakuta Y, Matsui I, Takabatake Y, Rakugi H, Shimizu S, Takahara S (2009) Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant Proc 41:52–54
Chien CT, Shyue SK, Lai MK (2007) Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 84:1183–1190
Kher A, Meldrum KK, Wang M, Tsai BM, Pitcher JM, Meldrum DR (2005) Cellular and molecular mechanisms of sex differences in renal ischemia-reperfusion injury. Cardiovasc Res 67:594–603
Kang KP, Lee JE, Lee AS, Jung YJ, Kim D, Lee S, Hwang HP, Kim W, Park SK (2014) Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol Med Rep 9:2061–2068
Schiebler TH, Danner KG (1978) The effect of sex hormones on the proximal tubules in the rat kidney. Cell Tissue Res 192:527–549
Rubinsztein DC, Marino G, Kroemer G (2011) Autophagy and aging. Cell 146:682–695
Reutzel-Selke A, Jurisch A, Denecke C, Pascher A, Martins PN, Kessler H, Tamura A, Utku N, Pratschke J, Neuhaus P, Tullius SG (2007) Donor age intensifies the early immune response after transplantation. Kidney Int 71:629–636
Decuypere JP, Pirenne J, Jochmans I (2014) Autophagy in renal ischemia-reperfusion injury: friend or foe? Am J Transplant 14:1464–1465
Bizargity P, Schroppel B (2014) Autophagy: basic principles and relevance to transplant immunity. Am J Transplant 14:1731–1739
Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM (2010) Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transplant 25:2187–2194
Belibi F, Zafar I, Ravichandran K, Segvic AB, Jani A, Ljubanovic DG, Edelstein CL (2011) Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am J Physiol Ren Physiol 300:F1235–F1243
Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, Pei Y, Musco G, Boletta A (2013) Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19:488–493
Cebotaru V, Cebotaru L, Kim H, Chiaravalli M, Boletta A, Qian F, Guggino WB (2014) Polycystin-1 negatively regulates polycystin-2 expression via the aggresome/autophagosome pathway. J Biol Chem 289:6404–6414
Gahl WA, Thoene JG, Schneider JA (2002) Cystinosis. N Engl J Med 347:111–121
Levtchenko E, Monnens L (2006) Development of Fanconi syndrome during infancy in a patient with cystinosis. Acta Paediatr 95:379–380
Wilmer MJ, Christensen EI, van den Heuvel LP, Monnens LA, Levtchenko EN (2008) Urinary protein excretion pattern and renal expression of megalin and cubilin in nephropathic cystinosis. Am J Kidney Dis 51:893–903
Ivanova E, De Leo MG, De Matteis MA, Levtchenko E (2014) Cystinosis: clinical presentation, pathogenesis and treatment. Pediatr Endocrinol Rev 12(Suppl 1):176–184
Kogan K, Spear ED, Kaiser CA, Fass D (2010) Structural conservation of components in the amino acid sensing branch of the TOR pathway in yeast and mammals. J Mol Biol 402:388–398
Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C (2005) The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell 19:15–26
Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ, Sarwal MM (2010) Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 21:272–283
Sansanwal P, Sarwal MM (2010) Abnormal mitochondrial autophagy in nephropathic cystinosis. Autophagy 6:971–973
Sansanwal P, Sarwal MM (2012) p62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis. Pediatr Nephrol 27:2137–2144
Sansanwal P, Li L, Sarwal MM (2015) Inhibition of intracellular clusterin attenuates cell death in nephropathic cystinosis. J Am Soc Nephrol 26:612–625
Napolitano G, Johnson JL, He J, Rocca CJ, Monfregola J, Pestonjamasp K, Cherqui S, Catz SD (2015) Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med 7:158–174
Acknowledgments
Stéphanie De Rechter and Elena Levtchenko are supported by the Fund for Scientific Research, Flanders 11M5214N, Fundamental Clinical Investigatorship 1801110N and has been supported by an IWT-SBO project (130033). Djalila Mekahli is supported by the Clinical Research Fund of UZ Leuven. Jean-Paul Decuypere is supported by a post-doctoral fellowship of ERA-EDTA.
Conflict of interest
The authors declare that they have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Answers to questions
1. D
2. B
3. E
4. C
5. B and C
Rights and permissions
About this article

Cite this article
De Rechter, S., Decuypere, JP., Ivanova, E. et al. Autophagy in renal diseases. Pediatr Nephrol 31, 737–752 (2016). https://doi.org/10.1007/s00467-015-3134-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-015-3134-2