
Abstract
Rationale
Many N-methyl-d-aspartate (NMDA) antagonists produce phencyclidine (PCP)-like side effects that limit their clinical utility. NMDA glycine-site antagonists may be less likely to produce these effects than other site-selective NMDA antagonists.
Objectives
The objective of the study is to compare the discriminative stimulus effects of novel NMDA glycine-site drugs to those of channel blocking and competitive NMDA antagonists.
Materials and methods
Drug discrimination studies were performed in separate groups of rats trained with saline vs. PCP (2 mg/kg i.p.) or the competitive antagonist NPC 17742 (4 mg/kg i.p.) using a standard two-lever operant conditioning procedure under an FR32.
Results
Neither the partial glycine-site agonists aminocyclopropane carboxylic acid methyl ester and (+)-HA-966 nor the antagonists L701,324; MDL 100,458; MDL 100,748; MDL 103,371; MDL 104,472; MDL 105,519; MRZ 2/571; MRZ 2/576; and ACEA 0762 produced >50% PCP-lever selection, though all were tested over a sufficient dose range to produce response rate decreasing effects. All of the antagonists, except MDL 100,458 and MDL 100,748, were also tested for NPC 17742-like effects, producing somewhat more variable results than in PCP-trained rats. ACEA-0762 produced full substitution for NPC 17742, whereas MDL 105,519 produced no substitution. The remaining compounds engendered between 20% and 80% drug-lever selection.
Conclusion
These results provide evidence that NMDA glycine-site partial agonists and antagonists generally do not produce discriminative stimulus effects similar to those of representative NMDA channel blockers or competitive antagonists. This suggests that these NMDA glycine-site antagonists should be less likely to produce the undesirable behavioral side effects seen in clinical trials with many other NMDA antagonists.
Similar content being viewed by others

Introduction
Excessive activation of the N-methyl-d-aspartate (NMDA) receptor has been linked to a number of acute and chronic nervous system disorders. Because of their potential for therapeutic use, numerous NMDA receptor antagonists are being developed for possible clinical use. The NMDA receptor has multiple ligand-binding sites through which ion channel opening can be modulated including the glutamate/NMDA binding site, the strychnine-insensitive glycine site (glycineB receptor), polyamine modulatory sites, and channel blocking sites. Binding of glycine concurrent with glutamate is required for activation of the receptor (Kleckner and Dingledine 1988), and thus, glycine is considered to be a co-agonist. The requirement for glycine binding in order to activate the ion channel allows manipulation of NMDA channel current by means of glycine-site ligands. Agonists, such as d-serine and d-cycloserine, have been investigated for use in enhancing cognitive function in dementias, particularly relating to memory and learning (Temple and Hamm 1996; Schwartz et al. 1996) as well as for treatment of schizophrenia (Wood 2005; Lipina et al. 2005). Partial agonists and antagonists have been investigated for many therapeutic uses including neuroprotection, analgesia, and anticonvulsant effects (Leeson and Iverson 1994; Danysz and Parsons 1998; Petty et al. 2003; Gigler et al. 2007). Interest in NMDA glycine-site antagonists is particularly strong because they have been predicted from animal studies to lack many phencyclidine (PCP)-associated side effects including neurodegenerative changes in the cingulate/retrosplenial cortex, learning impairment, and psychotomimetic effects (Berger et al. 1994; Balster et al. 1995, Witkin et al. 1997; Viu et al. 2000).
To further compare clinically relevant behavioral effects between site-selective NMDA antagonists and evaluate their potential for behavioral side effects, a variety of novel NMDA glycine-site drugs were evaluated for their ability to produce PCP- (representative channel blocking NMDA antagonist) and NPC 17742- (representative competitive NMDA antagonist) like discriminative stimulus effects in rats. Drug discrimination studies have been shown to be useful animal models for predicting subjective effects in humans (Holtzman 1990; Balster 1991). This procedure has been used to compare the behavioral effects of NMDA antagonists and has revealed differences among the discriminative stimulus effects of antagonists active at the various modulatory sites on the receptor complex. To date, the few glycine-site antagonists which have been tested appear to share little if any discriminative stimulus effects with either PCP or NPC 17742 (Singh et al. 1990b; Balster et al. 1995; Geter-Douglass and Witkin 1997; Witkin et al. 1997; Wiley et al. 1997; Beardsley et al. 2002) and thus would not be expected to produce the psychotomimetic effects that channel blocking and competitive NMDA antagonists have shown in humans (Dyker et al. 1999; Davis et al. 2000). The NMDA glycine-site antagonist drugs tested in the current study include a series of carboxyindole derivatives, two drugs from a series of tricyclic pyridophthalazine-diones and the highly potent and selective NMDA glycine-site antagonists, L701,324 and ACEA 0762. All of these compounds have been demonstrated to block NMDA ion channels in vitro as well as function as NMDA antagonists in one or more in vivo models (for review see Danysz and Parsons 1998). These compounds represent novel drugs purported to have higher CNS penetrability, greater potency, and/or increased selectivity for action at the NMDA glycine-site relative to many of the compounds tested in previous discrimination studies. In addition, two glycine-site partial agonists, (+)-HA-966 and the methyl ester of 1-aminocyclopropane-1-carboxylic acid (ACPCM), were also examined (Trullas et al. 1991; Priestley and Kemp 1994; Karcz-Kubicha et al. 1997).
Materials and methods
Virginia Commonwealth University is accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). All laboratory practices and animal care were consistent with current NIH guidelines, and all experimental protocols were approved by the Institutional Animal Care and Use Committee.
Procedure
Adult male Sprague–Dawley rats (Charles River, Wilmington, DE, USA) were trained to discriminate intraperitoneal (i.p.) injections of 2.0 mg/kg PCP or 4.0 mg/kg NPC 17742 from saline, as previously described (Willetts and Balster 1988; Wiley et al. 1997). They were individually housed with free access to water under a 12-h light/dark cycle. Food (Harlan Teklad Rodent Diet, Williamston, IL, USA) access beyond those obtained during behavioral sessions was restricted to approximately 15 g given postsession in order to increase lever-pressing for food. The subjects were trained daily (Monday–Friday) in 30-min sessions in standard two-lever operant conditioning chambers (Coulbourn Instruments, Lehigh Valley, PA, USA). Completion of a fixed ratio (FR) 32 on the correct lever resulted in delivery of a 45-mg food pellet (P.J. Noyes Company, Lancaster, NH, USA). The training drug (PCP or NPC 17742) and saline were given i.p. under a double alternation schedule (D, D, S, S, D, D, etc.) 15 min prior to session start for the PCP discrimination group and 1 h prior to session start for the NPC 17742 group. During sessions, a white stimulus light located centrally above each lever was illuminated. Incorrect responding reset the FR for correct-lever responding. Test sessions were conducted on Tuesday and Friday when the subjects met the following criteria on the preceding training sessions: (1) first FR completed on the correct lever, and (2) greater than 85% correct-lever responding over the entire session. During test sessions, completion of an FR on either lever resulted in the delivery of food reinforcement. Switching levers before completion of a consecutive FR resulted in resetting the FR count. Training continued under the double alternation of training drug and saline injections between test sessions. Illumination of lights, recording of responses and pellet delivery was performed using a SKED interface and SKED-11 operant conditioning software (State Systems, Kalamazoo, MI, USA) running on a DEC PCP-11BA23 minicomputer (Digital Equipment, Maynard, MA, USA).
Table 1 lists the NMDA sites of action, doses, and the vehicles for all test compounds. In addition to the glycine-site test drugs, a dose–effect curve for the appropriate training drug was obtained in each group of animals for purposes of comparison. All test drugs except those in dimethyl sulfoxide (DMSO) had injection volumes of 1 ml/kg. Drugs solubilized in DMSO (L 701,324 and ACEA 0762) had injection volumes of 0.5 ml/kg. The carboxyindole compounds (MDL compounds), (+)-HA966 and ACEA 0762 were administered 30 min presession; the tricyclic pyridophthalazine-diones (MRZ compounds) were given 20 min presession; L701,324 was administered 15 min presession, and ACPCM was given 10 min presession. The number of subjects tested with each compound varied and are detailed in the figure legends.
Data analysis
Data from test sessions were analyzed by determining the mean percentage of responses on the drug-associated lever and the overall mean response rate for all subjects. Data from sessions in which responding was less than 0.05 resp/s were excluded from determination of the mean percentage drug-lever responding, but all subject data were included in the response rate analyses. When less than three subjects qualified for inclusion, the data were not presented graphically. Full substitution for either training drug was defined as greater than 80% drug-lever responding. For comparing relative potencies in suppressing responding, the ED50 values for response rate effects (the dose required to cause a 50% decrease in rates of responding relative to saline control test rates) were calculated using regression analysis of the linear portions of the log 10 dose–effect curves. Control response rates were calculated for each subject as the average of the rates of responding on saline control tests that preceded and followed each dose–response curve determination. For test drugs that failed to decrease rates below 50%, the highest dose administered was used for comparison. Statistical analysis assessing the significance of response rate altering effects of drugs was conducted using one-way, within subject analysis of variance (SuperANOVA, Abacus Concepts, Berkeley, CA, USA). When the main effect of drug dose was significant (p < 0.05), post hoc Tukey LSD tests were performed to determine whether individual doses differed significantly (p < 0.05) from the saline control condition.
Drugs
PCP HCl was obtained from the National Institute on Drug Abuse (Rockville, MD, USA). NPC 17742 [2R,4R,5S-2-amino-4,5(1,2-cyclohexyl)-7-phosphonoheptanoic acid] was provided by NOVA Pharmaceutical (Baltimore, MD, USA). MDL 100,458 [3-(benzoylmethylamino)-6-chloro-1H-indole-2-carboxylic acid], MDL 100,748, MDL 103,371 [(E)-3-[2-(3-aminophenyl)-2-carboxyethenyl]-4,6-dichloro-1H-indole-2-carboxylic acid], MDL 104,472, and MDL 105,591[(E)-3-(2-phenyl-2-carboxyethenyl)-4,6-dichloro-1H-indole-2-carboxylic acid] were provided by Hoechst Marion Roussel (Cincinnati, OH, USA). MRZ 2/571[8-fluoro-4-hydroxy-1-oxo-1,2-dihydropyridazinol[4,5-b]quinoline-5-oxide (choline salt)] and MRZ 2/576 [8-chloro-4-hydroxy-1-oxo-1,2-dihydropyridazinol[4,5-b]quinoline-5-oxide (choline salt)] were obtained from Merz Pharmaceuticals GmbH (Frankfurt, Germany). L 701,324 [7-chloro-4-hydroxy-3(3-phenoxy)phenyl-2(1H)-quinolinone] was provided by Merck (Whitehouse, NJ, USA). CoCensys (Cranbury, NJ, USA) supplied the ACEA 0762 [5-aza-7-chloro-4-hydroxy-3-(m-phenoxyphenyl)quinoline-2(1H)-one]. (+)-HA-966 (3-amino-1-hydroxypyrrolid-2-one) was provided by Research Biochemicals International as part of the Chemical Synthesis Program of the National Institute of Mental Health Contract NOIMH00007.
Results
PCP-like effects
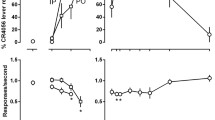
PCP produced a dose-dependent increase in PCP-lever responding with one or more doses producing full substitution (>80% PCP-lever selection) in all PCP-trained rats without concurrent response rate suppression (Figs. 1a, 2a, and 3a). Variable results were obtained with the NMDA glycine-site compounds, with none producing greater than 59% PCP-associated lever responding, and the intermediate levels of PCP-lever selection were generally associated with a decrease in rates of responding. MRZ 2/576 (Fig. 1a) produced a maximum of 33% PCP-lever selection at the 5.6 mg/kg dose; however, there was no dose dependency demonstrated as lower and higher doses of MRZ 2/576 failed to elicit any PCP-associated lever responding. The structurally similar compound MRZ 2/571 failed to produce PCP-associated lever responding at any dose tested. Both MRZ 2/571 [F 4,16 = 7.59, P = .001] and MRZ 2/576 [F 4,16 = 6.91, P = .002] were tested across similar dose ranges and produced significant dose-dependent decreases in rates of responding (Fig. 1a and Table 2).

Effects of the training drug, MRZ 2/571 and MRZ 2/576 in rats trained to discriminate 2 mg/kg PCP (a) or 4 mg/kg NPC 17742 (b) from saline. Shown in the upper graphs are mean (±SEM) percentage drug-lever responding and in the lower graphs are mean (±SEM) rates of responding. Values above PCP or NPC and Sal are the results of control tests with 2 mg/kg PCP or 4 mg/kg NPC 17742 and saline, respectively, conducted before testing each dose–effect curve. Mean percentage PCP-lever responding was based on five rats except for the following: 10 mg/kg MRZ 2/571 (n = 4); 5.6 and 10 mg/kg MRZ 2/576 (n = 3). Mean percentage NPC-lever responding was based on data from eight (NPC and MRZ 2/571) or six (MRZ 2/576) rats except for the following: 10 mg/kg MRZ 2/571 (n = 5); 10 and 17 mg/kg MRZ 2/576 (n = 4). Asterisks denote rates significantly different from saline (*p < 0.05, **p < 0.01)

Effects of the training drug, MDL 103,371, MDL 104,472, and MDL 105,519 in rats trained to discriminate 2 mg/kg PCP (a) or 4 mg/kg NPC 17742 (b) from saline. Shown in the upper graphs are mean (±SEM) percentage drug-lever responding and in the lower graphs are mean (±SEM) rates of responding. Values above PCP or NPC and Sal are the results of control tests with 2 mg/kg PCP or 4 mg/kg NPC 17742 and saline, respectively, conducted before testing each dose–effect curve. Values above Veh are the results from control tests conducted with 0.1% Tween-80 in each training group during testing of MDL 105,519. Mean percentage PCP-lever responding was based on data from seven (PCP and MDL 103,371) or six (MDL 104,472 and 105,519) rats except for the following: 8 mg/kg PCP (n = 4); 30 and 56 mg/kg MDL 104,472 (n = 3). The mean percentage PCP-lever responding for the MDL 105,519 doses of 30 and 56 mg/kg were each based on four of four rats. Mean percentage NPC-lever responding was based on data from seven (NPC and MDL 103,371), five (MDL 104,472), or four (MDL 105,519) rats except for the following: 16 mg/kg NPC 17742 (n = 6); 56 and 100 mg/kg MDL 103,371 (n = 6 and n = 3, respectively); 17 mg/kg MDL 104,472 (n = 4); 30 mg/kg MDL 105,519 (n = 3). Asterisks denote rates significantly different from saline (*p < 0.05, **p < 0.01)

Effects of the training drug, ACEA-0762, L701,324, and ACPCM (PCP group only) in rats trained to discriminate 2 mg/kg PCP (a) or 4 mg/kg NPC 17742 (b) from saline. Shown in the upper graphs are mean (±SEM) percentage drug-lever responding and in the lower graphs are mean (±SEM) rates of responding. Values above PCP or NPC and Sal are the results of control tests with 2 mg/kg PCP or 4 mg/kg NPC 17742 and saline, respectively, conducted before testing each dose–effect curve. Values above Veh are results for control tests with DMSO and 25% polyethylene glycol vehicles conducted during testing of ACEA 0762 and L 701,324. Mean percentage PCP-lever responding was based on data from 12 (L 701,324), seven (PCP and ACPCM), or five (ACEA 0762) rats except for the following: 8 mg/kg PCP (n = 4); 3 mg/kg ACEA 0762 (n = 3); 10 mg/kg L 701,324 (n = 11). Mean percentage NPC-lever responding was based on data from seven (NPC and ACEA 0762) or six (L 701,324) rats except for the following: 16 mg/kg NPC 17742 (n = 6); 3, 5.6, and 10 mg/kg L 701,324 (n = 5, 4, and 4, respectively); 3 and 5.6 mg/kg ACEA 0762 (n = 4 and 3, respectively). Asterisks denote rates significantly different from saline (*p < 0.05, **p < 0.01)
Testing of the carboxyindole derivative compounds produced a range of 0% to 59% PCP-associated lever selection (Fig. 2a and Table 2). The latter was produced by MDL 105,519 at the highest dose tested (100 mg/kg), but reflects the results for only two animals that were tested at this very high dose (data not presented graphically). One subject responded primarily on the PCP-associated lever and the other primarily on the saline-associated lever. Both subjects had low response rates relative to control levels. No other dose of MDL 105,519 elicited drug lever selection. MDL 103,371 and MDL 104,472 produced intermediate levels of PCP-associated lever responding at the highest doses tested. Both drugs produced a significant main effect on rates of responding [MDL103,371 F 5,30 = 3.02, P = .025; MDL 104,472 F 4,20 = 4.22, P = .012]. MDL 103,371 produced a maximum of 45% PCP-lever responding at 100 mg/kg accompanied by a 50% decrease in response rates, and MDL 104,472 produced a maximum level of 33% PCP-associated lever responding. Neither MDL 100,458 nor MDL 100,748 produced appreciable levels of PCP-associated lever selection across the dose ranges tested (Table 2).
The potent glycine site antagonist L 701,324 produced a maximum of 43% PCP-associated lever responding at 3 mg/kg with a significant dose-dependent decrease [F 4,44 = 7.61, P = .0001] to less than 20% PCP-lever responding as the dose was increased (Fig. 3a). This pattern of effect was demonstrated in two separate groups of PCP-trained rats (data combined in graph). On an individual subject basis, five of the 12 subjects demonstrated >90% PCP-lever selection at one or more doses without concomitant rate effects. Conversely, five subjects did not select the PCP-associated lever at any dose, and two subjects showed intermediate levels of lever selection accompanied by a >85% rate suppression. The partial agonist ACPCM also produced intermediate levels of PCP-associated lever responding at 300 mg/kg; however, this dose also produced a significant mean suppression of responding of over 60% [F 4,24 = 9.83, P = .0001] relative to control levels. ACEA 0762 (Fig. 3a) and the partial agonist (+)-HA966 (Table 2) both failed to elicit more than occasional responding on the PCP-associated lever even at doses significantly decreasing responding [ACEA0762 F 3,12 = 7.72, P = .004; HA966 F 4,20 = 36.88, P = .0001].
NPC 17742-like effects
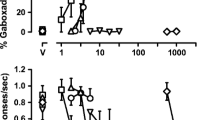
The competitive antagonist NPC 17742 produced a dose-dependent increase in NPC 17742-associated lever responding with one or more doses producing full substitution (>80%) in all NPC 17742-trained rats without concurrent response rate suppression (of Figs. 1b, 2b, and 3b). When tested in NPC17742-trained rats, the NMDA glycine-site compounds produced a wide range of drug-lever responding (<1% to 91%). MRZ 2/576 produced dose-dependent increases in NPC-associated lever responding (Fig. 1b), reaching a maximum of 69% at the highest dose tested (17 mg/kg) with accompanying decreases in response rates [F 4,24 = 13.20, P = .0001]. Both the ten (p < 0.05) and 17 (p < 0.01) mg/kg doses were associated with significant suppression of responding. Conversely, MRZ 2/571 occasioned only low levels of responding on the NPC 17742-associated lever, although it did produce significant rate effects[F 4,28 = 8.63, P = .0001].
The glycine-site antagonists MDL 103,371 and MDL 104,472 produced intermediate levels of NPC 17742-associated lever responding (Fig. 2b). MDL 103,371 produced a maximum of 52% substitution, and MDL 104,472 produced a maximum of 27% PCP-lever responding, but both occurred with concomitant response rate suppression [F 4,24 = 23.60, P = .0001], although only the MDL 103,371 dose of 100 mg/kg showed a significant decrease (p < 0.01). MDL 105,519 occasioned no appreciable responding on the NPC 17742-associated lever across the dose range tested.
ACEA-0762 produced >90% NPC 17742-associated lever responding at the highest dose tested (5.6 mg/kg); however, it also produced significant suppression of responding [F 4,24 = 8.62, P = .0002] with a 70% decrease in rates relative to saline control at the high dose (p < 0.01; Fig. 3b). L 701,324 produced intermediate levels of NPC 17742-associated lever responding reaching a peak of 46% at the highest dose tested (10 mg/kg). L 701,324 significantly reduced rate of responding [F 4,20 = 4.28, P = .0116] with 10 mg/kg producing a 75% decrease (p < 0.01) in rates relative to saline control levels (Fig. 3b).
Potency comparisons
The ED50 values for the test compounds to suppress responding are shown in Table 2. The relative order for suppression of responding in the PCP-trained rats was ACEA 0762 = MDL 100,748 > MRZ 2/576 > MRZ 2/571 > L 701,324 > MDL 104,472 > (+)-HA966 > MDL 103,371 > MDL 105,519 > MDL 100,458 ≫ ACPCM. The relative order for suppression of responding in the NPC 17742-trained rats was ACEA 0762 = L 701, 324 > MRZ 2/576 > MRZ 2/571 > MDL 104,472 > MDL 105,519 > MDL 103,371.
Discussion
Drug discrimination procedures have been used to compare and contrast the acute behavioral effects of site-selective NMDA antagonists demonstrating that differences exist based on site of action (for reviews see Willets et al. 1990; Balster 1991; Koek 1999). For example, in rats trained to discriminate PCP-like NMDA antagonists, competitive NMDA antagonists, such as NPC 17742 and CPP, produce at best only partial substitution for the training drug (Ferkany et al. 1989; Tricklebank et al. 1989; Koek et al. 1990; Nicholson and Balster 2003), while channel blockers such as ketamine and dizocilpine fully substitute (Balster 1991). When trained to discriminate competitive NMDA antagonists, rats and monkeys show complete substitution by NPC 17742 and other competitive antagonists, while PCP-like channel blockers show at most partial substitution accompanied by large decreases in response rates (Willetts et al. 1989; Bobelis and Balster 1993; Gold and Balster 1993; Wiley and Balster 1994). To date, only a few studies have trained discrimination of a glycine-site compound, and these have used either the low-efficacy partial agonist (+)-HA-966 (Singh et al. 1990b; Witkin et al. 1995) or its structural analog L-687,414 [R-(+)-cis-B-methyl-3-amino-1-hydroxypyrrolid-2-one] (Bourson and Tricklebank 1991). In these studies, partial agonists for the NMDA glycine-site fully substituted whereas the glycine-site antagonist 7-CK, channel blocking antagonists and NPC 17742 produced little if any substitution for the glycine-site compound.
In the current study, the NMDA glycine-site antagonists and partial agonists produced a range of PCP-lever selection but never resulted in greater than intermediate levels of PCP-lever responding (Figs. 1a, 2a, 3a). The antagonists MDL 100,458, MDL 100,748, MRZ 2/571, ACEA-0762, and the partial agonist (+)-HA-966 failed to produce substitution for PCP. These negative results are consistent with most previous studies testing NMDA glycine-site drugs. In rats trained to discriminate PCP or dizocilpine from saline, kynurenate, and 7-chlorokynurenic acid (7-CK), 5,7-dichlorokynurenic acid, (+)-HA-966, gavestinel [GV150526A; (E)-3[(phenylcarbamoil)ethenyl]-4,6-dichloroindole-2-carboxylic acid] and the isoquinolone compounds, ACEA 1011 (5-chloro-7-trifluoromethyl-1,4-dihydro-2,3-quinoxalinedione), ACEA 1021 (5-nitro-6,7-dichloro-1,4-dihydro-2,3-quinoxalinedione) all failed to produce even partial substitution for the training drug (Koek and Colpaert 1992; Singh et al. 1990b; Balster et al. 1995; Witkin et al. 1995, 1997; Beardsley et al. 2002). One weakness to the clinical utility of NMDA glycine-site antagonists has been their limited ability to cross the blood–brain barrier (Danysz and Parsons 1998). This may also limit their ability to function as central NMDA antagonists and may have been a component of the historically negative results seen when testing some NMDA glycine-site compounds. In the current study, all drugs that failed to engender PCP-lever responding above 20%, excluding MDL 100,458, were tested over a range which included at least one dose which significantly suppressed responding suggesting behaviorally active doses had been tested. Disruption of responding has typically been used as a nonspecific measure of behavioral effects and CNS penetration in discrimination studies. Because our procedure utilizes food reinforcement, however, it does not preclude the possibility of either anorectic or nauseating effects resulting in diminished responding. Therefore, it should be noted that these high doses also frequently resulted in grossly observable changes in behavior, including mild to strong sedation and motor ataxic effects. In addition, these compounds have all been shown to produce CNS effects following systemic administration in other behavioral procedures at or below the maximum doses tested in the current study (Baron et al. 1992; Millan and Sequin 1994; Kehne et al. 1995; Parsons et al. 1997).
In contrast to many earlier discrimination studies, the glycine-site antagonists MRZ 2/576 (Fig. 1a), L 701,324 (Fig. 3a), MDL 103,371, and MDL104,472 (Fig. 2a) as well as the partial agonist ACPCM (Fig. 3a), all produced a mean maximum percentage of PCP-lever responding between 33% and 45%. MDL 105,519 did elicit a mean of 59% PCP lever responding, but this represents data from only two subjects that were tested at the highest dose (100 mg/kg). Of these compounds, only L701,324 produced its maximum level of substitution without also producing ≥50% suppression of response rates. These intermediate levels of PCP-lever responding were accompanied by a high variability of lever selection between subjects, as reflected by the size of the error bars. Interpretation of intermediate (20% to 80%) levels of drug lever responding can be problematic. These levels of responding may reflect shared but incomplete overlap in the discriminative stimulus effects between the test drug and PCP consistent with partial substitution. Indeed, the levels of PCP-lever selection seen for MRZ 2/576, L701,324, and the three MDL compounds are not extremely different than that for many competitive NMDA antagonists in animals trained to discriminate PCP or PCP-like drugs. They are also consistent with results for 1-aminopropane carboxylic acid (ACPC), the parent compound of ACPCM (Koek and Colpaert 1992; Geter-Douglass and Witkin 1997). Thus, it may be, as we have increased the potency, selectivity, and/or CNS penetrability of NMDA glycine-site antagonists, we begin to see the production of more PCP-like behavioral effects. It is possible that if higher doses were tested, we might see even greater levels of PCP-lever responding. However, in consideration of the doses being used, relative to those producing therapeutic effects, the current procedure does provide information about behavioral effects over clinically relevant dose ranges. These compounds have been shown to produce desirable CNS effects, such as anti-seizure activity, neuroprotection, and antinociceptive effects at doses at or below those producing intermediate levels of PCP-lever responding (Baron et al. 1997; Bristow et al. 1995; Parsons et al. 1997; Lufty et al. 1997). Alternatively, the response rate suppression and gross behavioral effects seen when testing the NMDA glycine-site compounds and the high doses required to produce even intermediate levels of PCP-lever responding must be taken into consideration when interpreting these levels of drug lever responding. When disruption of responding is severe, it is possible that some level of PCP-lever selection reflects a nonspecific disruption of the PCP-saline discrimination and may not necessarily be an indication of PCP-like effects (Koek et al. 1993, 1999). This is the probable explanation for results seen when testing 100 mg/kg MDL 105,519. In these instances, the pattern of responding is more similar to that for GABAA-positive modulators in PCP-trained subjects than competitive NMDA antagonists (compare Browne 1982; Mansbach and Balster 1991 with Ferkany et al. 1989; Tricklebank et al. 1989; Koek et al. 1990; Nicholson and Balster 2003).
The results of substitution tests for NPC 17742, other than those for ACEA 0762, were in many ways similar to the substitution levels seen in the PCP-trained subjects. Previous testing of NMDA glcyine-site drugs in competitive NMDA antagonist-trained animals while limited to (+)-HA-966 and ACEA 1021, failed to elicit significant levels of substitution for the competitive antagonist (Wiley et al. 1997). Consistently, MDL 105,519 and MRZ 2/571 failed to substitute for NPC 17742. However, in the current study, MDL 103,371, MDL 104,472, MRZ 2/576, and L 701,324 did produce intermediate levels of NPC 17742-lever selection, and ACEA 0762 fully substituted for the NPC 17742 training dose which stands in sharp contrast to earlier studies. The greatest levels of substitution were all associated with decreases in rates of responding that were statistically significant except for MDL 104,472. The current data may reflect the behavioral effects of more potent compounds with greater CNS penetration, such that we begin to see intermediate or higher levels of substitution.
Potency estimates were calculated for the response rate-decreasing effects of the NMDA glycine-site drugs in both the PCP and NPC 17742 studies (Table 2). There was a wide range of potencies exhibited, with ACEA 0762, MDL 100,748, MRZ 2/576, and L701,324 being the most potent (ED50 values <10 mg/kg) and the partial agonist ACPCM being the least potent. There was generally a good correspondence between the potency for response rate effects in the two studies, although L701,324 was over three times more potent in the NPC 17742-trained rats than in the PCP-trained rats. The relative potencies to produce rate suppression did not correlate with binding affinity (compare current data to Baron et al. 1992; Kehne et al. 1995; Priestley et al. 1996; Lufty et al. 1997 and Danysz et al. 2005). However, given the variability between these compounds in their ability to access the CNS (Parsons et al. 1997; Hesselink et al. 1999), it is not an unexpected result.
When comparing among the glycine-site compounds, we see a range of behavioral effects, from compounds producing no PCP- or NPC-like effects (i.e., MRZ2/571), to those showing intermediate levels of training drug-lever responding in both groups (i.e., MDL 103,371) to production of >80% drugs lever responding seen for ACEA 0762 in the NPC 17742–trained group. While the NMDA glycine-site binding affinity, level of efficacy, and CNS penetration have been shown to vary between these drugs, all have produced effects associated with NMDA antagonist activity in vivo. Differences might be expected when comparing partial agonists to antagonists, but obvious differences were present between individual glycine-site antagonists, and similarities were observed between antagonists and partial agonists. For example, the tricyclic pyridophthalazine-dione MRZ 2/576 produced partial PCP- and NPC 17742-like discriminative stimulus effects, while the structurally similar MRZ 2/571 failed to substitute for either training drug. Yet, both of these compounds have been demonstrated to have similar anti-seizure effects at similar doses (Parsons et al. 1997). MDL 103,371 and MDL104,472 produced partial PCP-like discriminative stimulus effects, but the other carboxyindole derivatives did not. The antagonist L 701,324 and partial agonist ACPCM produced intermediate levels of PCP-lever responding, while the partial agonist (+)-HA-966 produced virtually none, despite its extremely low (∼13%) intrinsic activity (Singh et al. 1990a). The finding that not all site-selective compounds produce equivalent discriminative stimulus effects is not unique to the NMDA glycine co-agonist site. While drug discrimination results for high-affinity channel blockers have been fairly consistent, testing of low-affinity channel blockers has produced a wide spectrum of results from relatively high levels of PCP-like activity with memantine and dextromethorphan to those that completely lack PCP-like discriminative stimulus effects, such as ADCI [(±)-5-aminocarbonyl-10,11-dihydro-5H-dibenzo-[a,d]cyclohepten-5,10-imine] and amantadine (Grant et al. 1996; Nicholson et al. 1998; Nicholson and Balster 2002). In addition, differences in behavioral effects have been noted between NMDA glycine-site compounds, including between structurally similar compounds (Kehne et al. 1995). Thus, it is not unusual to see variable behavioral effects across these glycine-site compounds. It is possible that pharmacodynamic differences exist between these drugs, such as selectivity for NMDA receptor subpopulations, intrinsic activity, and binding kinetics, which may result in dissimilar behavioral profiles. It may also be that the divergent behavioral effects seen are due to non-NMDA receptor activity. While most of these compounds are considered selective for NMDA receptor binding, given variation in CNS penetration and other pharmacokinetic effects, additional CNS sites may play a role in the acute behavioral effects of some of these drugs.
In summary, a variety of different chemical families of NMDA glycine-site antagonists were tested for production of PCP-like or NPC 17742-like discriminative stimulus effects. Similar to many previous studies, some of the compounds failed to produce appreciable levels of drug-lever responding in either training group. In contrast, low to intermediate levels of substitution for PCP and NPC 17742 were observed for many of the compounds, suggesting potential overlap of discriminative stimulus effects between NMDA glycine-site drugs and the training drugs. However, these levels of substitution were seen primarily at very high doses, and most were accompanied by significant behavioral disruption, thereby distinguishing the acute behavioral effects of NMDA glycine-site antagonists in rats from the other site-selective NMDA antagonists. The current results also demonstrate a diversity of behavioral effects within the NMDA glycine-site drugs themselves. To understand the differences between the various NMDA glycine-site compounds, additional studies utilizing L701,324-trained rats have been initiated. The current results continue to suggest that many NMDA glycine-site antagonists should be less likely to produce the untoward behavioral side effects seen in clinical trials with various competitive and channel-blocking NMDA antagonists, particularly at clinically relevant doses.
References
Balster RL (1991) Discriminative stimulus properties of phencyclidine and other NMDA antagonists. In: Glennon RA, Järbe TUC, Frankenheim J (eds) Drug discrimination: Applications to drug abuse research, National Institute on Drug Abuse Research Monograph Series 116, DHHS Publication No.(ADM) 92-1878. U.S. Government Printing Office, Washington DC, pp 163–180
Balster RL, Mansbach RS, Shelton KL, Nicholson KL, Grech DM, Wiley JL, Li H, Weber E (1995) Behavioral pharmacology of two novel substituted quinoxalinedione glutamate antagonists. Behav Pharmacol 6:577–589
Baron BM, Harrison BL, McDonald IA, Meldrum BS, Palfreyman MG, Salituro FG, Siegel BW, Slone AL, Turner JP, White HS (1992) Potent indole- and quinoline-containing N-methyl-d-aspartate antagonists acting at the strychnine-insensitive glycine binding site. J Pharmacol Exp Ther 262:947–956
Baron BM, Harrison BL, Kehne JH, Schmidt CJ, van Giersbergen PL, White HS, Siegel BW, Senyah Y, McCloskey TC, Fadayel GM, Taylor VL, Murawsky MK, Nyce P, Salituro FG (1997) Pharmacological characterization of MDL 105,519, an NMDA receptor glycine site antagonist. Eur J Pharmacol 323:181–192
Beardsley PM, Ratti E, Balster RL, Willetts J, Trist D (2002) The selective glycine antagonist gavestinel lacks phencyclidine-like behavioral effects. Behav Pharmacol 13:583–592
Berger P, Farrel K, Sharp F, Skolnick P (1994) Drugs acting at the strychnine-insensitive glycine receptor do not induce HSP-70 protein in the cingulate cortex. Neurosci Lett 168:147–150
Bobelis DJ, Balster RL (1993) Pharmacological specificity of the discriminative stimulus properties of 2-amino-4,5-(1,2-cyclohexyl)-7-phosphonoheptanoic acid (NPC 12626), a competitive N-methyl-d-aspartate receptor antagonist. J Pharmacol Exp Ther 264:845–853
Bourson A, Tricklebank MD (1991) The discriminative stimulus properties of the glycine/NMDA receptor antagonist L-687,414. Fund Clin Pharmacol 5:443
Bristow LJ, Landon L, Saywell KL, Tricklebank MD (1995) The glycine/NMDA antagonist, L-701,324 reverses isolation-induced deficits in prepulse inhibition in the rat. Psychopharmacology 118:230–232
Browne RG (1982) Discriminative stimulus properties of phencyclidine. In: Colpaert FC, Slangen JL (eds) Drug discrimination: applications in CNS pharmacology. Elsevier, Amsterdam, pp 109–122
Danysz W, Parsons CG (1998) Glycine and N-methyl-d-aspartate receptors: Physiological significance and possible therapeutic applications. Pharmacol Rev 50:597–664
Danysz W, Kozela E, Parsons CG, Sladek M, Bauer T, Popik P (2005) Peripherally acting NMDA receptor/glycineB site receptor antagonists inhibit morphine tolerance. Neuropharmacology 48:360–371
Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, Norris J (2000) Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke 31:347–354
Dyker AG, Edwards KR, Fayad PB, Hormes JT, Lees KR (1999) Safety and tolerability study of aptiganel hydrochloride in patients with an acute ischemic stroke. Stroke 30:2038–2042
Ferkany JW, Kyle DJ, Willetts J, Rzeszotarski WJ, Guzewska ME, Ellenberger SR, Jones SM, Sacaan AI, Snell LD, Borosky S, Jones BE, Johnson KM, Balster RL, Burchett K, Kawasaki K, Hoch DB, Dingledine R (1989) Pharmacological profile of NPC 12626, a novel, competitive N-methyl-D-aspartate receptor antagonist. J Pharmacol Exp Ther 250:100–109
Geter-Douglass B, Witkin JM (1997) Dizocilpine-like discriminative stimulus effects of competitive NMDA receptor antagonists in mice. Psychopharmacology 133:43–50
Gigler G, Szénási G, Simó A, Lévay G, Hársing LG Jr, Sas K, Vécsei L, Toldi J (2007) Neuroprotective effect of l-kynurenine sulfate administered before focal cerebral ischemia in mice and global cerebral ischemia in gerbils. Eur J Pharmacol 564:116–122
Gold LH, Balster RL (1993) Effects of NMDA receptor antagonists in squirrel monkeys trained to discriminate the competitive NMDA receptor antagonist NPC 12626 from saline. Eur J Pharmacol 230:285–292
Grant KA, Colombo G, Grant J, Rogawski MA (1996) Dizocilpine-like discriminative stimulus effects of low-affinity uncompetitive NMDA antagonists. Neuropharmacology 35:1709–1719
Hesselink MB, Smolders H, Eilbacher B, De Boer AG, Breimer DD, Danysz W (1999) The role of probenecid-sensitive organic acid transport in the pharmacokinetics of N-methyl-d-aspartate receptor antagonists acting at the glycine(B)-site: microdialysis and maximum electroshock seizures studies. J Pharmacol Exp Ther 290:543–550
Holtzman SG (1990) Discriminative stimulus effects of drugs: relationship to potential for abuse. In: Adler MA, Cowan A (eds) Testing and evaluation of drugs of abuse. Wiley-Liss, New York, pp 193–210
Karcz-Kubicha M, Jessa M, Nazar M, Plaznik A, Hartmann S, Parsons CG, Danysz W (1997) Anxiolytic activity of glycine-B antagonists and partial agonists—no relation to intrinsic activity in the patch clamp. Neuropharmacology 36:1355–1367
Kehne JH, Baron BM, Harrison BL, McCloskey TC, Palfreyman MG, Poirot M, Salituro FG, Siegel BW, Slone AL, Van Giersbergen PL et al (1995) MDL 100,458 and MDL 102,288: two potent and selective glycine receptor antagonists with different functional profiles. Eur J Pharmacol 284:109–118
Kleckner NW, Dingledine R (1988) Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241:835–837
Koek W (1999) N-methyl-d-aspartate antagonists and drug discrimination. Pharmacol Biochem Behav 64:275–281
Koek W, Colpaert FC (1992) N-methyl-d-aspartate antagonism and phencyclidine-like activity: behavioral effects of glycine site ligands. In: Kamenka JM, Domino EF (eds) Multiple sigma and PCP receptor ligands: mechanisms for neuromodulation and neuroprotection. NPP Books, Ann Arbor, Michigan, pp 655–671
Koek W, Woods JH, Colpaert FC (1990) N-methyl-d-aspartate antagonism and phencyclidine-like activity: a drug discrimination analysis. J Pharmacol Exp Ther 253:1017–1025
Koek W, Colpaert FC, Vignon J (1993) Effects of phencyclidine-type drugs in rats discriminating fentanyl from saline: pharmacological and behavioral characterization of intermediate levels of drug lever selection. J Pharmacol Exp Ther 264:746–756
Leeson PD, Iverson LL (1994) The glycine site on the NMDA receptor: structure–activity relationships and therapeutic potential. J Med Chem 37:4053–4067
Lipina T, Labrie V, Weiner I, Roder J (2005) Modulators of the glycine site on NMDA receptors, d-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology 179:54–67
Lufty K, Cai SX, Woodward RM, Weber E (1997) Antinociceptive effects of NMDA and non-NMDA receptor antagonists in the tail flick test in mice. Pain 70:31–40
Mansbach RS, Balster RL (1991) Pharmacological specificity of the phencyclidine discriminative stimulus in rats. Pharmacol Biochem Behav 39:971–975
Millan MJ, Seguin L (1994) Chemically-diverse ligands at the glycine B site coupled to N-methyl-d-aspartate (NMDA) receptors selectively block the late phase of formalin-induced pain in mice. Neurosci Lett 178:139–143
Nicholson KL, Balster RL (2002) Evaluation of the discriminative stimulus effects of the low-affinity N-methyl-d-aspartate channel blockers AR-R 13950AA and AR-R 16283AA in rats and rhesus monkeys. Behav Pharmacol 13:571–581
Nicholson KL, Balster RL (2003) Evaluation of the phencyclidine-like discriminative stimulus effects of novel NMDA channel blockers in rats. Psychopharmacoogy 170:215–224
Nicholson KL, Jones HE, Balster RL (1998) Evaluation of the reinforcing and discriminative stimulus properties of the low-affinity N-methyl-d-aspartate channel blocker memantine. Behav Pharmacol 9:231–243
Parsons CG, Danysz W, Quack G, Hartmann S, Lorenz B, Wollenburg C, Baran L, Przegalinski E, Kostowski W, Krzascik P, Chizh B, Headley PM (1997) Novel systemically active antagonists of the glycine site of the N-methyl-d-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther 283:1264–1275
Petty MA, Neumann-Haefelin C, Kalisch J, Sarhan S, Wettstein JG, Juretschke HP (2003) In vivo neuroprotective effects of ACEA 1021 confirmed by magnetic resonance imaging in ischemic stroke. Eur J Pharmacol 474:53–62
Priestley T, Kemp JA (1994) Kinetic study of the interactions between the glutamate and glycine recognition sites on the N-methyl-d-aspartic acid receptor complex. Mol Pharmacol 46:1191–1196
Priestley T, Laughton P, Macaulay AJ, Hill RG, Kemp JA (1996) Electrophysiological characterisation of the antagonist properties of two novel NMDA receptor glycine site antagonists, L-695,902 and L-701,324. Neuropharmacology 35:1573–1581
Schwartz BL, Hashtroudi S, Herting RL, Schwartz P, Deutsch SI (1996) d-Cycloserine enhances implicit memory in Alzheimer patients. Neurology 46:420–424
Singh L, Donald AE, Foster AC, Hutson PH, Iversen LL, Iversen SD, Kemp JA, Leeson PD, Marshall GR, Oles RJ et al (1990a) Enantiomers of HA-966 (3-amino-1-hydroxypyrrolid-2-one) exhibit distinct central nervous system effects: (+)-HA-966 is a selective glycine/N-methyl-d-aspartate receptor antagonist, but (−)-HA-966 is a potent gamma-butyrolactone-like sedative. Proc Natl Acad Sci 87:347–351
Singh L, Menzies R, Tricklebank MD (1990b) The discriminative stimulus properties of (+)-HA-966, an antagonist at the glycine/N-methyl-d-aspartate receptor. Eur J Pharmacol 186:129–132
Temple MD, Hamm RJ (1996) Chronic, post-injury administration of D-cycloserine, an NMDA partial agonist, enhances cognitive performance following experimental brain injury. Brain Res 741:246–251
Tricklebank MD, Singh L, Oles RJ, Preston C, Iversen SD (1989) The behavioural effects of MK-801: a comparison with antagonists acting non-competitively and competitively at the NMDA receptor. Eur J Pharmacol 167:127–135
Trullas R, Folio T, Young A, Miller R, Boje K, Skolnick P (1991) 1-aminocyclopropanecarboxylates exhibit antidepressant and anxiolytic actions in animal models. Eur J Pharmacol 203:379–385
Viu E, Zapata A, Capdevila J, Skolnick P, Trullas R (2000) Glycine(B) receptor antagonists and partial agonists prevent memory deficits in inhibitory avoidance learning. Neurobiol Learn Mem 74:146–160
Witkin JM, Brave S, French D, Geter-Douglass B (1995) Discriminative stimulus effects of R-(+)-3-amino-1-hydroxypyrrolid-2-one, [(+)-HA-966], a partial agonist of the strychnine-insensitive modulatory site of the N-methyl-D-aspartate receptor. J Pharmacol Exp Ther 275:1267–1273
Witkin JM, Steele TD, Sharpe LG (1997) Effects of strychnine-insensitive glycine receptor ligands in rats discriminating dizocilpine or phencyclidine from saline. J Pharmacol Exper Ther 280:46–52
Wiley JL, Balster RL (1994) Effects of competitive and non-competitive N-methyl-d-aspartate (NMDA) antagonists in squirrel monkeys trained to discriminate D-CPPene (SDZ EAA 494) from vehicle. Psychopharmacology 116:266–272
Wiley JL, Li H, Balster RL (1997) Discriminative stimulus effects of site-selective N-methyl-d-aspartate antagonists in NPC 17742-trained rats and squirrel monkeys. Psychopharmacology 132:382–388
Willetts J, Balster RL (1988) The discriminative stimulus effects of N-methyl-d-aspartate antagonists in phencyclidine-trained rats. Neuropharmacology 27:1249–1256
Willetts J, Bobelis DJ, Balster RL (1989) Drug discrimination based on the competitive N-methyl-d-aspartate antagonist, NPC 12626. Psychopharmacology 99:458–462
Willetts J, Balster RL, Leander JD (1990) The behavioral pharmacology of NMDA receptor antagonists. Trends Pharmacol Sci 11:423–428
Wood PL (2005) The NMDA receptor complex: a long and winding road to therapeutics. Idrugs 8:229–235
Acknowledgment
The technical assistance of Hua Li is greatly appreciated. Special thanks to Dr. Linda Bristow of Merck, Sharpe & Dohme Laboratories for the supply of L 701,324; Drs. Chris Parsons and Wojciech Danysz at Merz & Co. for the supply of MRZ 2/576 and MRZ 2/571; Dr. Bruce Baron of Hoechst Marion Roussel for supplying the MDL compounds; Dr. Richard Carter of CoCensys, Inc. for supplying ACEA 0762; and Dr. J. Willetts of NOVA Pharmaceuticals for supplying NPC 17742.
This research was supported by NIDA grant R01DA-01442.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nicholson, K.L., Balster, R.L. The discriminative stimulus effects of N-methyl-d-aspartate glycine-site ligands in NMDA antagonist-trained rats. Psychopharmacology 203, 441–451 (2009). https://doi.org/10.1007/s00213-009-1469-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1469-8