
Abstract
Aims/hypothesis
Incretin-based therapies improve glycaemic control in patients with type 2 diabetes. In animal models of diabetes, glucagon-like peptide-1 receptor agonists (GLP-1RAs) increase beta cell mass. GLP-1RAs are also evaluated in non-diabetic individuals with obesity and cardiovascular disease. However, their effect on beta cell mass in normoglycaemic conditions is not clear. Here, we investigate the effects of the GLP-1RA liraglutide on beta cell mass and function in normoglycaemic mice.
Methods
C57BL/6J mice were treated with the GLP-1RA liraglutide or PBS and fed a control or high-fat diet (HFD) for 1 or 6 weeks. Glucose and insulin tolerance tests were performed after 6 weeks. BrdU was given to label proliferating cells 1 week before the animals were killed. The pancreas was taken for either histology or islet isolation followed by a glucose-induced insulin-secretion test.
Results
Treatment with liraglutide for 6 weeks led to increased insulin sensitivity and attenuation of HFD-induced insulin resistance. A reduction in beta cell mass was observed in liraglutide-treated control and HFD-fed mice at 6 weeks, and was associated with a lower beta cell proliferation rate after 1 week of treatment. A similar reduction in alpha cell mass occurred, resulting in an unchanged alpha to beta cell ratio. In contrast, acinar cell proliferation was increased. Finally, islets isolated from liraglutide-treated control mice had enhanced glucose-induced insulin secretion.
Conclusions/interpretation
Our data show that GLP-1RA treatment in normoglycaemic mice leads to increases in insulin sensitivity and beta cell function that are associated with reduced beta cell mass to maintain normoglycaemia.
Similar content being viewed by others


Introduction
Glucagon-like peptide 1 (GLP-1) is an incretin hormone secreted by intestinal L cells in response to ingestion of carbohydrates and lipids [1]. Activation of the GLP-1 receptor (GLP-1R) on pancreatic beta cells leads to glucose-dependent insulin secretion and improves glycaemic control in patients with type 2 diabetes [2, 3]. In animal models of diabetes these therapies increase beta cell mass [4–6].
The beta cell mass is tightly controlled in order to keep glucose levels within a narrow range. When the demand for insulin is chronically increased by physiological or pathological changes, such as pregnancy or obesity, there is an increase in beta cell function and/or beta cell mass [7, 8]. When the demand for insulin decreases, for example postpartum, the beta cell mass reverts to its original capacity [9].
Besides their effects on beta cells, GLP-1R agonists (GLP-1RAs) exert several extrapancreatic effects that may be of therapeutic benefit. GLP-1RAs decrease body weight and are associated with reduced blood pressure, improved lipid profiles and improved endothelial function in patients with type 2 diabetes [10, 11]. Therefore, these compounds have also been evaluated in non-diabetic individuals with obesity or cardiovascular disease [12–15]. However, the effect of GLP-1RAs on beta cells in these normoglycaemic conditions is not clear. Therefore, we investigated the effects of GLP-1RA treatment on beta cell mass and function in normoglycaemic mice.
Methods
Animals
Male C57BL/6J mice, 8–9 weeks old (Charles River Laboratories, Wilmington, MA, USA), were housed under standard conditions with a 12 h light/dark cycle and free access to food and water. Mice were fed a normal diet (control; 10% of total energy intake derived from lard fat, 16.3 kJ [3.9 kcal]/g; D12450B, Research Diets, New Brunswick, NJ, USA) or a high-fat diet (HFD; 45% of total energy intake derived from lard fat, 19.7 kJ [4.7 kcal]/g; D12451, Research Diets) for 1 or 6 weeks. Average food intake was determined weekly per cage housing three or four mice. Liraglutide (0.1 mg/kg, Novo Nordisk, Bagsvaerd, Denmark) or PBS was given twice daily with at least a 10 h interval between subcutaneous injections. Treatment was discontinued 1 day before the animals were killed. Body weight was determined after overnight fasting. Animal experiments were approved by the institutional ethical committee on animal care and experimentation at the Leiden University Medical Center.
Glucose and insulin tolerance tests
Insulin tolerance and glucose tolerance were assessed after 6 weeks of treatment. An intraperitoneal insulin tolerance test (ITT) was performed in animals that had been fasted for 6 h. After measuring basal blood glucose concentration from the tail vein, 0.75 U/kg insulin was injected followed by monitoring of the blood glucose concentrations after 15, 30 and 60 min. An intraperitoneal glucose tolerance test (GTT) was performed in overnight-fasted mice. Blood samples were drawn from the tail vein before injecting 2 g/kg glucose and after 15, 30, 60 and 120 min. Blood glucose concentrations were measured using a glucose meter (Accu-Chek, Roche, Basel, Switzerland) and insulin concentrations were measured by ELISA (Chrystal Chem, Downers Grove, IL, USA). Plasma IL-6, IL-1β and monocyte chemoattractant protein-1 (MCP-1) were detected using a custom cytokine/metabolic multiplex assay (Meso Scale Discovery, Gaithersburg, MD, USA).
Pancreas dissection and islet isolation
Mice were anaesthetised by isoflurane inhalation and exsanguinated. The pancreases were dissected and weighed. For immunohistochemistry the pancreas was fixed by immersion in a 4% (vol./vol.) paraformaldehyde solution. For islet isolation the pancreases of six to eight mice were pooled and digested using 3 mg/ml collagenase (Sigma-Aldrich, St Louis, CA, USA) in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2 μg/ml DNAse I (Pulmozyme, Roche) and shaken at 37°C for 15–18 min until a homogeneous digest was obtained. The digest was then washed three times with cold RPMI medium supplemented with 10% (vol./vol.) heat-inactivated FCS (Bodinco, Alkmaar, the Netherlands) and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively; Invitrogen). Islets were purified from exocrine tissue by manual selection picking under a dissecting microscope.
Glucose-induced insulin secretion test
A glucose-induced insulin secretion test was performed on freshly isolated islets. Groups of ten islets were incubated in a modified Krebs-Ringer bicarbonate buffer with HEPES (KRBH) containing 115 mmol/l NaCl, 5 mmol/l KCl, 24 mmol/l NaHCO3, 2.2 mmol/l CaCl2, 1 mmol/l MgCl2, 20 mmol/l HEPES and 2 g/l human serum albumin (Cealb, Sanquin, the Netherlands), pH 7.4. Islets were washed and pre-incubated with KRBH buffer containing 2 mmol/l glucose for 1.5 h at 37°C. They were then incubated in 2 mmol/l glucose KRBH buffer for 1 h at 37°C and switched to 20 mmol/l glucose KRBH buffer for 1 h at 37°C. Supernatant fractions were kept for determination of insulin concentration by ELISA (Mercodia, Uppsala, Sweden). Islet cells were lysed by sonication in distilled water. Islet insulin content was measured by acid ethanol extraction followed by ELISA (Mercodia). Islet DNA content was determined by Quant-iT PicoGreen dsDNA kit (Invitrogen).
Immunohistochemistry and morphometry
In order to obtain representative samples of the entire organ, pancreases from each mouse (six per group) were cut into three pieces (duodenal, gastric and splenic region) [16] that were embedded in paraffin blocks and sliced into 4 μm sections. For each analysis two to four sections per block, with an interval of at least 200 μm between sections, were immunostained and analysed. The average of the three regions was taken as a measure for the entire organ.
For the identification of beta cells, nine to twelve sections per mouse pancreas were immunostained with guinea pig anti-insulin IgG (Millipore, Billerica, MA, USA) or rabbit anti-insulin IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h followed by horseradish peroxidase (HRP)- or alkaline phosphatase-conjugated secondary antibodies for 1 h. Alpha cells were identified by immunostaining by rabbit anti-glucagon IgG (Vector Laboratories, Burlingame, CA, USA) for 1 h followed by HRP-conjugated secondary antibody for 1 h. Sections were developed with 3,3′-diaminobenzidine tetrahydrochloride (DAB) or Liquid Permanent Red (LPR; Dako, Glostrup, Denmark) and counterstained with haematoxylin. Stained sections were digitally imaged (Panoramic MIDI, 3DHISTECH, Budapest, Hungary).
Beta cell and pancreas areas stained with haematoxylin were determined using an image-analysis program (Stacks 2.1, LUMC, Leiden, the Netherlands), excluding large blood vessels, larger ducts, adipose tissue and lymph nodes as previously described [16]. Beta cell mass was determined by the ratio of beta cell area to pancreas area multiplied by the pancreas weight. Beta cell cluster area was determined as the average area of beta cell clusters (defined as ≥4 beta cells per cluster) per mouse. Alpha cell mass was determined by calculating the ratio of alpha cell area to beta cell area per islet, using ImageJ software (ImageJ, US National Institutes of Health, Bethesda, MD, USA), multiplied by the beta cell mass.
To label proliferating beta cells, mice were given 50 mg/kg BrdU (Sigma-Aldrich) subcutaneously twice daily during the entire period for the 1 week study and the final 7 days for the 6 week study. Sections were double stained for insulin-LPR and BrdU (BrdU staining kit, Invitrogen). Stained sections were digitally imaged (Panoramic MIDI). BrdU-positive beta cells were assessed as a proportion of all beta cells. Pancreatic duct cells were identified based on their typical morphology and location. The number of BrdU-positive duct cells was counted. The number of BrdU-positive acinar cells was counted using Stacks 2.1 and expressed as a percentage of the total number of acinar cells. The area in which these were counted was divided by the total number of cells as a measure of acinar cell size. Apoptotic beta cells were identified by the TUNEL technique (Roche) in combination with insulin immunostaining and were counted. The investigator was blind to the experimental conditions during counting.
Statistical analysis
Data are presented as mean±SEM. Statistical calculations were carried out using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). The statistical significance of differences was determined by two-way ANOVA, followed by Bonferroni’s multiple comparisons test, as appropriate. p < 0.05 was considered statistically significant.
Results
Metabolic characteristics of control and HFD-fed mice following 6 weeks of liraglutide treatment
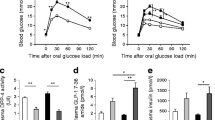
Liraglutide treatment for 6 weeks was associated with decreased body weight and increased insulin sensitivity in both control and HFD-fed mice (Fig. 1a–c). After a glucose load, liraglutide treatment attenuated the peak glucose concentration induced by HFD at 30 min (Fig. 1d), but this did not reach significance for the AUC for glucose (Fig. 1e). In mice on a normal diet there was no significant difference in glucose concentrations after liraglutide treatment (Fig. 1d). In both liraglutide-treated groups the early response of insulin secretion during the GTT was increased to a similar extent as in HFD-fed mice (Fig. 1f, g).

Metabolic characteristics of control (squares) and HFD-fed (circles) mice treated with liraglutide (black) or PBS (white) for 6 weeks. (a) Body weight (n = 13–14 mice). (b) Blood glucose concentrations expressed as percentage of basal glucose concentration during the ITT (n = 7–8 mice). (c) AUC of glucose concentrations during the ITT corrected for basal glucose concentration (n = 7–8 mice). (d) Blood glucose concentrations during GTT (n = 6 mice). (e) AUC for blood glucose concentrations during the GTT (n = 6 mice). (f) Insulin concentrations during GTT (n = 5–6 mice). (g) AUC 0–15 min insulin concentrations during GTT (n = 5–6 mice). *p < 0.05, **p < 0.01 and ***p < 0.001; † p < 0.05 vs control + PBS; ‡ p < 0.05 vs control + PBS and HFD + liraglutide
Increased acinar cell proliferation after liraglutide treatment
After 6 weeks, pancreatic weight was significantly greater in liraglutide-treated mice (Fig. 2a), despite the decrease in body weight (Fig. 1a). Therefore, we analysed the effect of liraglutide treatment on the exocrine pancreas. Treatment with liraglutide was associated with an increased size of acinar cells in control mice (Fig. 2b). We counted 13,441 ± 439 acinar cells per mouse. The number of proliferating acinar cells was 65% higher in liraglutide-treated control mice (Fig. 2c, d). A similar effect was observed in liraglutide-treated HFD-fed mice, though the difference was less prominent. No significant difference in the number of proliferating duct cells was observed between the groups after 6 weeks of treatment (528 ± 28 duct cells were counted per mouse; Fig. 2e, f). Assessment of pro-inflammatory cytokine plasma concentrations (IL-1β, IL-6 and MCP-1) showed no significant differences between the groups (electronic supplementary material [ESM] Fig. 1).

The effects of liraglutide (black bars) or PBS (white bars) treatment on the exocrine pancreas in control and HFD-fed mice after 6 weeks. (a) Pancreas weight (n = 13–14 mice). (b) Acinar cell size (n = 6 mice). (c) Representative picture of proliferating acinar cells, BrdU (brown); scale bar, 100 μm. (d) Acinar cell proliferation, BrdU labelling during the final 7 days (n = 6 mice). (e) Duct cell proliferation, BrdU labelling during the final 7 days (n = 6 mice). (f) Representative picture of proliferating duct cells (arrows), BrdU (brown); scale bar, 50 μm. *p < 0.05, **p < 0.01 and ***p < 0.001
Liraglutide reduces beta cell and alpha cell mass
Beta cell mass, determined by analysing 94.9 ± 4.7 mm2 pancreatic tissue per mouse, was reduced in control and HFD-fed mice after treatment with liraglutide for 6 weeks (Fig. 3a). This was associated with a decreased average beta cell cluster area (Fig. 3b). Liraglutide treatment did not affect insulin content in freshly isolated islets (Fig. 3c). No difference in the number of apoptotic beta cells was found between groups after counting 1,129 ± 170 beta cells per mouse (Fig. 3d). To determine the number of proliferating beta cells 1,807 ± 128 cells per mouse were counted. Liraglutide treatment in both control and HFD-fed mice was associated with a lower number of proliferating beta cells after 1 week, but after 6 weeks no difference was observed (Fig. 3e–g). Similarly, the alpha cell mass was reduced in liraglutide-treated control mice. The lower alpha cell mass in HFD-fed mice was unaffected by liraglutide treatment (Fig. 3h). The ratio of alpha to beta cell area did not change significantly between the groups (Fig. 3i).

Beta cell and alpha cell mass in control and HFD-fed mice after liraglutide (black bars) or PBS (white bars) treatment. (a) Beta cell mass after 6 weeks of treatment (n = 6 mice). (b) Beta cell cluster area after 6 weeks of treatment (n = 6 mice). (c) Insulin content from isolated islets corrected for DNA content (n = 24). (d) Beta cell apoptosis, identified by TUNEL+/insulin+ staining, after 6 weeks of treatment (n = 6). (e) Beta cell proliferation in control mice after 1 and 6 weeks of treatment, BrdU labelling during the final 7 days (n = 6 mice). (f) Beta cell proliferation in HFD-fed mice after 1 and 6 weeks of treatment, BrdU labelling during the final 7 days (n = 6 mice). (g) Representative picture of proliferating beta cells (arrows), BrdU (brown) and insulin (red). Scale bar = 50 μm. (h) Alpha cell mass after 6 weeks of treatment (n = 6 mice). (i) Ratio of alpha cell area to beta cell area after 6 weeks of treatment (n = 6 mice). *p < 0.05, **p < 0.01 and ***p < 0.001
Increased glucose-induced insulin release from isolated islets of liraglutide-treated control mice
Finally, in order to investigate whether 6 weeks of liraglutide treatment had specific effects on beta cell function in the presence of a reduced beta cell mass, we assessed glucose-induced insulin secretion in isolated islets. Basal insulin secretion (2 mmol/l glucose) was increased twofold in islets from liraglutide-treated control mice (Fig. 4a). Glucose stimulation of islets from liraglutide-treated control mice resulted in a 35% increase in insulin secretion (Fig. 4a). As expected, glucose stimulation of islets from HFD-fed mice led to increased insulin secretion compared with mice fed regular chow (Fig. 4b). This glucose stimulation was unchanged in HFD-fed mice that had been treated with liraglutide for 6 weeks (Fig. 4b).

Glucose-induced insulin secretion from isolated islets of control mice and HFD-fed mice treated with liraglutide for 6 weeks. Insulin secretion is presented as a percentage of total insulin content. (a) Insulin secretion of islets from control mice, n = 24, and (b) insulin secretion of islets from HFD-fed mice, n = 23–24; white bars, 2 mmol/l glucose; grey bars, 20 mmol/l glucose. *p < 0.05, **p < 0.01 and ***p < 0.001
Discussion
The effect of GLP-1-based therapy on insulin secretion from beta cells has been reported to be glucose dependent [17–19], but its effect on beta cell mass under different glycaemic conditions is less clear. While GLP-1RA treatment increases beta cell mass in animal models of diabetes, we now show a reduction in beta cell mass in normoglycaemic mice.
GLP1-RA treatment of non-diabetic obese individuals results in weight loss and improved beta cell function [12, 13]. Therefore, it is relevant to understand how beta cell mass adapts during GLP1-RA treatment under normoglycaemic conditions and different dietary situations. So far, few studies have investigated the effect of GLP-1RA in non-diabetic animals and showed either no difference or an increase in beta cell proliferation after short-term treatment for between 2 and 10 days [6, 20–22].
Liraglutide treatment for 6 weeks resulted in decreased body weight and increased insulin sensitivity in normoglycaemic mice and HFD-fed mice, in line with earlier studies [23, 24]. In addition, the increased early response of insulin secretion during the GTT in liraglutide-treated mice is in line with the working mechanism of GLP-1 [25, 26]. This was associated with a major reduction in beta cell mass in both control and HFD-fed mice. The lower beta cell proliferation rate that we observed in mice treated for 1 week suggests that the beta cell mass adapted rapidly after the start of liraglutide treatment. Interestingly, we also show that alpha cell mass was reduced to such an extent that the ratio of alpha to beta cells remained unchanged.
Acute GLP-1R stimulation of beta cells is known to increase insulin secretion in a glucose-dependent manner [17–19]. We show that sustained GLP-1RA treatment during normoglycaemic conditions is associated with increased insulin secretion from isolated islets even in the absence of direct GLP-1RA stimulation in vitro. In HFD-fed mice, liraglutide treatment increased insulin sensitivity, but the enhanced insulin secretory response remained. Together these data suggest that liraglutide treatment in normoglycaemic mice leads to increased insulin sensitivity and an enhanced insulin secretory response from existing beta cells, thereby reducing the need for new beta cells. In addition, these data imply that chronic GLP-1RA treatment during normoglycaemia results in an increased beta cell function as was shown in non-diabetic obese individuals [12]. In contrast, GLP-1RA treatment does not increase beta cell proliferation during normoglycaemia.
Finally, we observed increased acinar cell proliferation after liraglutide treatment indicating that this effect can occur during normoglycaemia. There was a non-significant difference in duct cell proliferation after 6 weeks of treatment. These observations and the findings of other studies [27–32] raise the issue of whether GLP-1-based therapies are a potential risk for pancreatitis; some studies did not observe this effect [33, 34], however, which may reflect the animal model used, the age of the animals and/or the labelling and counting methods used for proliferating cells. In our study, there was no indication of a systemic inflammatory state. Ductal proliferation is also associated with the development of pancreatic adenocarcinoma but the relationship between GLP-1-based treatment and the development of new pancreatic malignant lesions is not clear [35].
In conclusion, our data indicate that GLP-1RA under normoglycaemic conditions can have different effects on pancreatic islet and non-islet cells. GLP-1RA treatment during normoglycaemia results in a reduction in beta cell mass, whereas it exerts proliferative effects on the exocrine pancreas.
Abbreviations
- GLP-1:
-
Glucagon-like peptide-1
- GLP-1R:
-
Glucagon-like peptide-1 receptor
- GLP-1RA:
-
Glucagon-like peptide-1 receptor agonist
- GTT:
-
Glucose tolerance test
- HFD:
-
High-fat diet
- HRP:
-
Horseradish peroxidase
- ITT:
-
Insulin tolerance test
- KRBH:
-
Krebs-Ringer bicarbonate buffer with HEPES
- LPR:
-
Liquid Permanent Red
- MCP-1:
-
Monocyte chemoattractant protein-1
References
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439
Blonde L, Russell-Jones D (2009) The safety and efficacy of liraglutide with or without oral antidiabetic drug therapy in type 2 diabetes: an overview of the LEAD 1-5 studies. Diabetes Obes Metab 11(Suppl 3):26–34
Vilsboll T (2009) The effects of glucagon-like peptide-1 on the beta cell. Diabetes Obes Metab 11(Suppl 3):11–18
Farilla L, Hui H, Bertolotto C et al (2002) Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 143:4397–4408
Rolin B, Larsen MO, Gotfredsen CF et al (2002) The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am J Physiol Endocrinol Metab 283:E745–E752
Xu G, Stoffers DA, Habener JF, Bonner-Weir S (1999) Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 48:2270–2276
Bonner-Weir S (2000) Perspective: postnatal pancreatic beta cell growth. Endocrinology 141:1926–1929
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Rieck S, Kaestner KH (2010) Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab 21:151–158
Vilsboll T, Christensen M, Junker AE, Knop FK, Gluud LL (2012) Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ 344:d7771
Ussher JR, Drucker DJ (2012) Cardiovascular biology of the incretin system. Endocr Rev 33:187–215
Astrup A, Rossner S, van Gaal L et al (2009) Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 374:1606–1616
Rosenstock J, Klaff LJ, Schwartz S et al (2010) Effects of exenatide and lifestyle modification on body weight and glucose tolerance in obese subjects with and without pre-diabetes. Diabetes Care 33:1173–1175
Nikolaidis LA, Mankad S, Sokos GG et al (2004) Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 109:962–965
Fields AV, Patterson B, Karnik AA, Shannon RP (2009) Glucagon-like peptide-1 and myocardial protection: more than glycemic control. Clin Cardiol 32:236–243
Ellenbroek JH, Tons HA, de Graaf N et al (2013) Topologically heterogeneous beta cell adaptation in response to high-fat diet in mice. PLoS One 8:e56922
Kreymann B, Williams G, Ghatei MA, Bloom SR (1987) Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 2:1300–1304
Meloni AR, Deyoung MB, Lowe C, Parkes DG (2013) GLP-1 receptor activated insulin secretion from pancreatic beta-cells: mechanism and glucose dependence. Diabetes Obes Metab 15:15–27
Weir GC, Mojsov S, Hendrick GK, Habener JF (1989) Glucagonlike peptide I (7-37) actions on endocrine pancreas. Diabetes 38:338–342
Edvell A, Lindstrom P (1999) Initiation of increased pancreatic islet growth in young normoglycemic mice (Umea +/?). Endocrinology 140:778–783
Fan R, Kang Z, He L, Chan J, Xu G (2011) Exendin-4 improves blood glucose control in both young and aging normal non-diabetic mice, possible contribution of beta cell independent effects. PLoS One 6:e20443
Bock T, Pakkenberg B, Buschard K (2003) The endocrine pancreas in non-diabetic rats after short-term and long-term treatment with the long-acting GLP-1 derivative NN2211. APMIS 111:1117–1124
Young AA, Gedulin BR, Bhavsar S et al (1999) Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48:1026–1034
Parlevliet ET, de Leeuw van Weenen JE, Romijn JA, Pijl H (2010) GLP-1 treatment reduces endogenous insulin resistance via activation of central GLP-1 receptors in mice fed a high-fat diet. Am J Physiol Endocrinol Metab 299:E318–E324
Mojsov S, Weir GC, Habener JF (1987) Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest 79:616–619
Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF (1987) Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA 84:3434–3438
Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC (2011) Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 141:150–156
Koehler JA, Baggio LL, Lamont BJ, Ali S, Drucker DJ (2009) Glucagon-like peptide-1 receptor activation modulates pancreatitis-associated gene expression but does not modify the susceptibility to experimental pancreatitis in mice. Diabetes 58:2148–2161
Nachnani JS, Bulchandani DG, Nookala A et al (2010) Biochemical and histological effects of exendin-4 (exenatide) on the rat pancreas. Diabetologia 53:153–159
Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC (2012) Chronic GLP-1 receptor activation by exendin-4 induces expansion of pancreatic duct glands in rats and accelerates formation of dysplastic lesions and chronic pancreatitis in the Kras(G12D) mouse model. Diabetes 61:1250–1262
Perfetti R, Zhou J, Doyle ME, Egan JM (2000) Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology 141:4600–4605
Singh S, Chang HY, Richards TM, Weiner JP, Clark JM, Segal JB (2013) Glucagon-like peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: a population-based matched case-control study. JAMA Intern Med 73:534–539
Nyborg NC, Molck AM, Madsen LW, Knudsen LB (2012) The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes 61:1243–1249
Vrang N, Jelsing J, Simonsen L et al (2012) The effects of 13 wk of liraglutide treatment on endocrine and exocrine pancreas in male and female ZDF rats: a quantitative and qualitative analysis revealing no evidence of drug-induced pancreatitis. Am J Physiol Endocrinol Metab 303:E253–E264
Vangoitsenhoven R, Mathieu C, van der Schueren B (2012) GLP1 and cancer: friend or foe? Endocr Relat Cancer 19:F77–F88
Funding
This work was supported by the Diabetes Cell Therapy Initiative consortium, which includes the Dutch Diabetes Research Foundation, and an unrestricted research grant from Novo Nordisk.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
JHE contributed to the conception and design of the study and the acquisition, analysis and interpretation of the data and drafted/revised the article. HAMT, MJAWvM, NdG and MAH contributed to the acquisition of the data and drafted/revised the article. TJR, FC and EJPdK contributed to the conception and design of the study, the analysis and interpretation of the data and drafted/revised the article. All authors gave final approval of the manuscript to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 69 kb)
Rights and permissions
About this article
Cite this article
Ellenbroek, J.H., Töns, H.A.M., Westerouen van Meeteren, M.J.A. et al. Glucagon-like peptide-1 receptor agonist treatment reduces beta cell mass in normoglycaemic mice. Diabetologia 56, 1980–1986 (2013). https://doi.org/10.1007/s00125-013-2957-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2957-2