
Abstract
Hypoglycaemic agents such as sulphonylureas and the newer group of "glinides" stimulate insulin secretion by closing ATP-sensitive potassium (KATP) channels in pancreatic beta cells, but have varying cross-reactivity with related channels in extrapancreatic tissues such as heart, vascular smooth and skeletal muscle. Experiments on the structure-function relationships of recombinant KATP channels and the phenotypes of mice deficient in different KATP channel subunits have provided important insights into the mechanisms underlying sulphonylurea selectivity, and the potential consequences of KATP channel blockade outside the pancreatic beta cell. The different pharmacological properties of KATP channels from beta cells compared with those from cardiac, smooth and skeletal muscle, are accounted for by the expression of alternative types of sulphonylurea receptor, with non-identical drug binding sites. The sulphonylureas and glinides are found to fall into two groups: one exhibiting selectivity for beta cell sulphonylurea receptors (SUR1), and the other blocking cardiovascular and skeletal muscle sulphonylurea receptors (SUR2) with potencies similar to their action on SUR1. In seeking potential side effects of KATP channel inhibitors in humans, it is essential to take these drug differences into account, along with the probability (suggested by the studies on KATP channel knockout mice) that the effects of extrapancreatic KATP channel inhibition might be either subtle or rare. Further studies are still required before a final decision can be made on whether non-selective agents are appropriate for the therapy of Type 2 diabetes.
Similar content being viewed by others


Recent years have witnessed extensive progress in our understanding of the physiology and pharmacology of ATP-sensitive potassium (KATP) channels. The cloning of the sulphonylurea receptor (SUR1) in 1995 [1], and soon afterwards of its binding partner, the inwardly rectifying potassium channel Kir6.2 [2, 3], opened the way for an integrated investigation into KATP channel structure and function, combining techniques drawn from molecular biology, electrophysiology, biochemistry and transgenic/knockout technology (reviewed in [4, 5, 6]). As a result, we are now beginning to recognise the physiological roles of KATP channels in a range of tissues, and the molecular basis for the observed variation in their pharmacological properties.
Therapeutic modulation of beta cell KATP channel activity has been used for many years to enhance insulin release in subjects with Type 2 diabetes [4, 5, 6]. Sulphonylureas and the newer class of 'glinides' stimulate insulin secretion by interacting with SUR1, thereby bringing about KATP channel closure and increased electrical activity in the beta cells. However, KATP channels have been described in a number of extrapancreatic tissues, including cardiac, skeletal and smooth muscle and some neurones [4, 5, 6, 7, 8, 9, 10, 11, 12, 13], and are blocked to varying degrees by the different sulphonylureas and glinides used in clinical practice. beta cell-selective and non-selective agents are generally used interchangeably in the treatment of diabetes, because studies have not conclusively shown that blocking extrapancreatic KATP channels has a deleterious effect. As we are now beginning to understand the physiological roles of non-beta cell KATP channels and the molecular basis for tissue-selective KATP channel inhibition, perhaps the time has come to revisit this issue. This review will therefore focus on the molecular mechanisms of action of sulphonylureas and related drugs, and the possible consequences of non-selective KATP channel inhibition, based on the knowledge we have gained from studies on recombinant KATP channels and KATP channel-deficient mice.
The unrivalled role of KATP channels in beta cell physiology
Although KATP channels have been identified in a range of muscular, neuronal and neuroendocrine tissues, their function is best understood in pancreatic beta cells where they are involved in coupling membrane electrical events and insulin release to changes in the plasma glucose concentration [14, 15]. The release of insulin-containing secretory granules is triggered by an increase in the local cytoplasmic Ca2+ concentration, and occurs when Ca2+ enters the beta cell through Ca2+ channels in the surface membrane. The opening of these channels is controlled by the membrane potential, which is itself determined by the balance between the inward and outward currents (Fig. 1). At low glucose concentrations outward currents are dominant, due to the high rate of K+ efflux through open KATP channels. The net efflux of positively charged ions sets up a membrane potential of about –70 mV (inside negative). At high glucose levels the KATP channels close, the magnitude of the K+ current falls below that of the inward currents, and the membrane potential drifts to less negative values (depolarisation). Voltage-gated Ca2+ channels open as the membrane depolarises, allowing Ca2+ entry and the stimulation of insulin secretion.

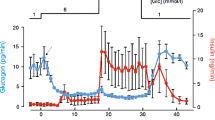
KATP channel activity modulates insulin secretion in pancreatic beta cells. Glucose-dependent KATP channel closure results in membrane depolarisation and insulin release, which are further enhanced by the addition of arginine
A principal determinant of KATP channel activity is believed to be the intracellular concentrations of the adenine nucleotides, ATP and ADP. These nucleotides have opposing actions on KATP channels, with ATP acting as a channel blocker, and MgADP as a channel opener [4, 5, 6]. In pancreatic beta cells, the metabolic rate is responsive to the plasma glucose concentration, resulting in enhanced glycolytic and mitochondrial generation of ATP from ADP at high glucose levels [16, 17, 18]. The increase in ATP and concomitant fall in ADP concentration therefore combine to close the KATP channels, triggering Ca2+ entry and insulin release.
The glucose-dependent reduction in K+ efflux also enables beta cells to respond to small inward currents, such as that generated by arginine uptake (Fig. 1). Transport of positively charged arginine molecules by the cationic amino acid transporter is electrogenic, i.e. there is a net charge movement across the cell membrane associated with arginine uptake, producing a small depolarising current [19]. Arginine-stimulated insulin release is glucose-dependent because the transporter current is very small and is drowned by the opposing KATP current at low glucose concentrations. It only becomes an important player when the KATP channels are largely closed (at raised glucose levels), and can then enhance depolarisation and insulin release. Although it is not known whether other electrogenic transporters play any role in beta cells, sodium glucose cotransporters in GLP-1 secreting cells exert a similar effect, providing a glucose-dependent inward current that enhances depolarisation and GLP-1 release following KATP channel closure [20, 21].
Molecular architecture of KATP channels
The K+ conducting pore of KATP channels is formed from four inwardly-rectifying potassium (Kir) channel subunits, either Kir6.1 or Kir6.2 [2, 3, 22, 23, 24]. Kir6.2 forms the KATP channel pore in the majority of tissues, but expression of the alternative pore subunit, Kir6.1, e.g. in vascular smooth muscle and astrocytes, generates KATP channels with different pore properties and nucleotide sensitivity [25, 26, 27]. Kir subunits have intracellular N and C termini, and two transmembrane helices separated by a pore loop (Fig. 2). Isolated Kir6.2 tetramers are retained in the endoplasmic reticulum by an Arg-Lys-Arg (RKR) motif located in the C-terminus, but are trafficked to the surface membrane on co-assembly with sulphonylurea receptors [28]. Kir6.2 channels can also be engineered to reach the surface membrane without SUR by truncation of the RKR-containing C-terminus, a feature that has been exploited to investigate the intrinsic properties of the Kir6.2 subunit [29]. One of the most notable properties of truncated Kir6.2 subunits, expressed without SUR, is that they are blocked by adenine nucleotides (ATP>ADP) [29, 30].

Membrane topology of Kir6.2 and SUR1 subunits. The transmembrane and intracellular domains of SUR1 and Kir6.2 are indicated. The cytoplasmic linkers are depicted as separate intracellular domains, by analogy with the structure of MsbA [163]. Regions implicated in the binding and action of sulphonylureas are shown in grey, and the position of residue S1237 is indicated by a white circle. TMD = transmembrane domain; NBD = nucleotide binding domain
In fully-assembled KATP channels, each of the four Kir6 subunits is associated with a regulatory sulphonylurea receptor (Fig. 2) [23, 24]. SURs originate from two separate genes and occur in several alternatively-spliced isoforms. SUR1 has been identified in pancreatic beta and alpha cells, GLP-1 secreting intestinal L-cells, and neurones (e.g. in the ventromedial and mediobasal hypothalamus, substantia nigra and myenteric nervous system) [1, 12, 20, 31, 32, 33]. SUR2 is predominantly found in muscle, and exists in two important alternatively-spliced isoforms: SUR2A in skeletal and cardiac muscle, and SUR2B in smooth muscle [25, 34, 35]. Although additional splice variations of both SUR1 and SUR2 have been described, their physiological roles remain unclear [36, 37, 38, 39, 40]. SURs are members of the ATP binding cassette (ABC) transporter family. They have 17 transmembrane helices (TMs 1–17) arranged in three domains (TMD0, TMD1 and TMD2), containing five, six and six helices, respectively. They also possess two large intracellular nucleotide binding domains (NBDs), the first located between TMDs 1 and 2, and the second lying distal to TMD2 [41, 42].
The sulphonylurea receptors endow KATP channels with sensitivity to the stimulatory actions of Mg-nucleotides and KATP channel openers (e.g. diazoxide, nicorandil) and the inhibitory effects of sulphonylureas and glinides [4, 5, 29, 34, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56]. Their NBDs contain consensus nucleotide binding motifs—the Walker A and Walker B motifs—which, in other ABC transporters, are the sites of MgATP binding and hydrolysis [58, 59]. In SUR, binding of MgADP at NBD2 is believed to underlie nucleotide-dependent channel activation [60, 61, 62, 63], and it has been suggested that ADP shuttling from mitochondria to KATP channels might be facilitated by enzymes such as adenylate kinase and creatine kinase, which have been found to form physical associations with SUR2 [64, 65]. In biochemical studies, however, it has also been shown that the NBDs of SUR can hydrolyse MgATP [62, 63, 66, 67, 68]. This would generate MgADP at NBD2 and presumably therefore promote channel opening, raising the possibility that channel activity could also be regulated in vivo by physiological modulation of the ATPase activity of NBD2 [62].
Human KATP channel mutations associated with hyperinsulinism and diabetes
A number of polymorphisms in Kir6.2 and SUR1 subunits have been detected in the human population. Many have no effect on KATP channel function, but others have been associated with hyperinsulinism and diabetes. The first pathogenic mutations in SUR1 to be discovered were in subjects with congenital hyperinsulinism (CHI or persistent hyperinsulinaemic hypoglycaemia of infancy, PHHI) [56, 69]. This condition presents in early life with persistent life-threatening hypoglycaemia and raised insulin concentrations, and in the most severe cases is treatable only by sub-total pancreatectomy. Pancreatic beta cells from these subjects show a lack of KATP channel activity in the surface membrane, causing continuous membrane depolarisation, persistently raised intracellular calcium concentrations and unregulated insulin release [70]. Over 40 disease-causing mutations in SUR1, and 4 in Kir6.2 have been described, accounting for about 50% of the known cases of CHI [5, 6, 71, 72, 73]. Further characterisation of the SUR1 mutations has shown that they result in premature stop codons, trafficking defects or loss of KATP channel sensitivity to the endogenous activator, MgADP [56, 69, 71, 72, 74, 75, 76, 77]. The loss of MgADP sensitivity prevents KATP channels from opening during hypoglycaemia, and the association of such mutations with CHI supports the idea that the crucial signal opening KATP channels during metabolic stress is the nucleotide-dependent channel activation mediated via SUR1, rather than the falling ATP concentration [56].
A mutation in SUR1 (E1506K) that causes mild autosomal dominant CHI in infants, has also been found to cause autosomal dominant Type 2 diabetes in adult life [78, 79]. This mutation in NBD2 interferes with channel activation by MgADP, accounting for the CHI phenotype [78]. By puberty, however, a severe defect in glucose-stimulated insulin secretion could be measured in both normoglycaemic and diabetic carriers of the mutation [79]. The maintenance of normoglycaemia in many of these subjects indicates the physiological importance of KATP channel independent pathways in the beta cell such as incretin-induced secretion and vagal stimuli. The development of diabetes was associated with obesity in three out of four subjects, and probably resulted from an increased insulin requirement that could not be met by the failing beta cells. Although the mechanisms underlying beta cell failure in this group are not clear, an increased rate of apoptosis, perhaps as a result of the chronic membrane depolarisation, has been proposed. If this is true, it could have implications for chronic therapy with potent long-acting sulphonylureas, which might be predicted to induce a similar state of persistent beta cell depolarisation.
A number of recent studies have also demonstrated a strong association between Type 2 diabetes and the common KATP channel polymorphism, Kir6.2-E23K [80, 81, 82]. Functional studies revealed that recombinant KATP channels containing the Kir6.2-E23K mutation spend more time in the open state, and have reduced sensitivity to ATP inhibition and enhanced activation by MgGDP [83, 84]. In human physiological studies the E23K variant was associated with impaired insulin secretion and glucagon suppression during an OGTT and increased BMI, but normal insulin release in response to intravenous glucose [85, 86]. One possible explanation for the diabetic tendency might be that release of incretins in response to oral glucose is also impaired, because the KATP channels in GLP-1 secreting L-cells have the same molecular composition as those in beta cells [20]. Why the beta cells show apparently normal glucose sensitivity in vivo, despite the KATP channel defect, is unclear. Impaired secretion of the L-cell hormones, GLP-1 and Peptide YY, and alterations in hypothalamic KATP channel activity involved in feeding behaviour might also contribute to the development of obesity [86].
Sulphonylurea action
Drugs that bind to sulphonylurea receptors and close KATP channels have been used for many years to stimulate insulin release in subjects with Type 2 diabetes. Although the first and second generation agents (e.g. tolbutamide, chlorpropamide, glibenclamide, gliclazide, glimepiride) possess a sulphonylurea group, this is not essential for drug activity. Indeed, meglitinide and its derivatives, which are structurally related to the non-sulphonylurea half of glibenclamide, form a new class of therapeutic KATP channel inhibitors, the glinides. Drugs within this category (e.g. meglitinide, nateglinide, repaglinide and mitiglinide) do not, however, seem to interact with identical regions of the sulphonylurea receptor.
Studies comparing the binding and functional effects of different KATP channel inhibitors have been carried out using recombinant KATP channels expressed in Xenopus oocytes and mammalian cell lines. Channels formed by coexpression of Kir6.2 subunits with either SUR1, SUR2A or SUR2B, replicate the properties of native KATP channels from beta cells, cardiac and smooth muscle, respectively, and express at high levels. They are amenable to genetic manipulations, facilitating investigations into the molecular mechanisms of drug action. Measurement of the current flowing through patches of membrane taken from oocytes expressing Kir6.2/SUR complexes, provides a direct measure of the K+ flux and therefore of the number of open channels (typically thousands) in the patch of oocyte membrane. A reduction in the current is observed when agents close the KATP channels, mirroring the pharmacological action of the drugs in vivo [4].
With the first studies of tolbutamide action on Kir6.2/SUR1 currents in oocyte membranes, it became apparent that the dose inhibition curve reflected block at two independent binding sites with very different affinities (Fig. 3) [87]. Low concentrations of the drug, causing high-affinity inhibition, only reduced the current amplitude by 50 to 75%. A flat section of the curve was then followed by a second, low-affinity, component of block, occurring at much higher drug concentrations. The high-affinity component reflects drug binding to the sulphonylurea receptor and occurs in the range of concentrations used therapeutically. The low-affinity block is only observed at suprapharmacological drug levels, and has been attributed to direct interaction of the compound with the Kir6.2 subunit [87, 88]. Recognition of the low-affinity component has clarified the interpretation of dose response relationships that were previously fitted with a single shallow curve.

Dose response relationships for inhibition of beta cell and cardiac-type KATP currents by sulphonylureas and glinides. Kir6.2/SUR1 and Kir6.2/SUR2A currents are inhibited by increasing concentrations of sulphonylureas and glinides. In the upper row are shown drugs that inhibit SUR1-type channels with higher potency than SUR2-type channels. In the lower row are shown drugs that inhibit SUR1-type and SUR2-type channels with similar potencies. The conductance in the presence of drug (G) is shown relative to that in control solution (Gc). Experiments were performed in membrane patches excised from Xenopus oocytes expressing Kir6.2 with the SUR shown, and solutions did not contain nucleotides. SUR1, closed squares and solid lines; SUR2A, open circles and dashed lines. Data are taken from references [44, 45, 46, 47, 48]
Subsequent studies with a range of sulphonylureas and glinides confirmed that they all exhibit both high and low affinity components of inhibition, with a variable separation between the affinities of the two sites (Fig. 3) [44, 45, 46, 47, 48, 50]. Drug interaction with the SUR subunit always resulted in a maximum reduction in the current amplitude of 50 to 75%, when the agents were added on their own to the cytoplasmic surface of the membrane. The interpretation at a molecular level is that the channels remain able to open, albeit less readily, when drug is bound to the sulphonylurea receptor [89]. This behaviour is modified in the presence of cytoplasmic nucleotides, as discussed later.
Whereas all hypoglycaemic sulphonylureas and glinides were found to block SUR1-type channels with high affinity, however, they fell into two distinct groups when their effects on SUR2-type channels were compared (Fig. 3) [44, 45, 46, 47, 48, 50]. Agents in the first group (glibenclamide, glimepiride, repaglinide, meglitinide) showed high affinity block of Kir6.2/SUR2 currents, with potencies similar to those observed with SUR1 subunits. Interestingly, these agents all possessed a non-sulphonylurea moiety. Drugs in the second group (tolbutamide, gliclazide, nateglinide) did not exhibit high-affinity block of Kir6.2/SUR2 currents, and only closed the channels at concentrations that could interact directly with the Kir6.2 subunit. Chlorpropamide is also SUR1-selective, as it inhibited Kir6.2/SUR1 currents by 75 ± 3% and Kir6.2/SUR2A currents by only 17±5% (n=5 of each) at a concentration of 100 µmol/l (five- to ten-fold above the Kd for chlorpropamide displacement of glibenclamide binding to beta cell membranes [90]) (Fig. 4).

Reversibility and structure of sulphonylureas and glinides. The reversibility of different sulphonylureas and glinides was measured on Kir6.2/SUR1 and Kir6.2/SUR2A currents expressed in Xenopus oocytes. Currents are shown in response to repeated voltage ramps from –110 to +100 mV, and the vertical bars represent 1 nA. Drugs were added as indicated, at the following concentrations: chlorpropamide (100 µmol/l), gliclazide (10 µmol/l), glibenclamide (100 nmol/l), glimepiride (100 nmol/l), meglitinide (10 µmol/l), repaglinide (1 µmol/l). The drug structures are indicated to the left. Data were taken from the following references: gliclazide [45], glibenclamide and meglitinide [44], repaglinide [47]
The ability of tolbutamide to inhibit Kir6.2/SUR1, but not Kir6.2/SUR2, channels with high affinity provided a pharmacological tool to search for the sulphonylurea binding site. Chimeric sulphonylurea receptors were constructed in which isolated domains were swapped between SUR1 and SUR2, to identify regions of SUR1 that are obligatory for the high affinity action of tolbutamide. In this way, the critical site was narrowed down to a section within the last TM domain (TMD2) of SUR1 (Fig. 2) [91]. A more detailed analysis of this area resulted in the identification of a single serine residue (S1237) in the cytoplasmic loop (CL8) linking TM helices 15 and 16, which, when mutated to its SUR2 counterpart (tyrosine), abolished both high-affinity tolbutamide block and [3H]glibenclamide binding [91]. Subsequent studies have confirmed the importance of this cytoplasmic loop, and of residue S1237, for sulphonylurea binding to SUR1 [49, 52]. Consistent with these data, introduction of the reverse mutation into SUR2B (Y1206S) increased the [3H]glibenclamide binding affinity about ten-fold [92]. As tyrosine has a more bulky side chain than serine, it is suggested that this in some way obstructs drug binding to SUR2.
When carrying out patch clamp experiments, it is evident that agents that block SUR1-type KATP channels with high affinity have marked differences in their rates of reversal (Fig. 4) [44, 45, 46, 47]. At a structural level, there is a close relationship between drug reversibility and the number of potential binding interactions with SUR1. Most drugs that possess only one active group (either a sulphonylurea or a non-sulphonylurea) are rapidly reversible over a recording period of less than 30 s. This group includes tolbutamide, chlorpropamide, gliclazide and meglitinide. By contrast, drugs that possess both sulphonylurea and non-sulphonylurea moieties (glibenclamide and glimepiride) are effectively irreversible in patch clamp experiments (measured over several minutes). As often occurs, there is an exception to the rule, in this case repaglinide, which does not possess a sulphonylurea group yet is very difficult to wash off KATP channels in membrane patches.
A similar idea holds for Kir6.2/SUR2 currents, but in this case the lack of binding of the sulphonylurea group to SUR2 results in a different pattern of reversibility (Fig. 4) [44, 45, 46, 47]. Thus, tolbutamide, chlorpropamide and gliclazide do not exhibit high-affinity block of Kir6.2/SUR2 currents, inhibition by meglitinide, glibenclamide and glimepiride probably only involves binding of a non-sulphonylurea moiety and is rapidly reversible, and block by repaglinide is effectively irreversible, as with SUR1.
The binding site for sulphonylureas and glinides could therefore be envisaged as a pocket with at least two binding motifs, one (exclusive to SUR1 and abolished by the S1237Y mutation) favouring sulphonylurea groups, and the other (common to SUR1 and SUR2) preferring meglitinide-like molecules [4, 44, 91]. Drug binding to more than one motif produces a particularly strong interaction, and is effectively irreversible. The slow reversibility of repaglinide inhibition might reflect additional interactions of the hydrophobic groups in this molecule. Slow reversibility seems to be crucial for the biochemical detection of drug binding, as specific binding to SUR1 and beta cell membranes can be measured with [3H] derivatives of glibenclamide, glimepiride and repaglinide, but not with [3H]nateglinide [1, 49, 90, 93, 94]. The difficulties sometimes encountered in trying to measure [3H]glibenclamide binding to SUR2-type channels and subunits [34] might be attributable to its rapid reversibility. In line with this idea, mutation of S1237Y in SUR1 increased glibenclamide reversibility in patch clamp experiments and impaired the binding of [3H]glibenclamide [49, 91], whereas the reverse mutation (Y1206S) in SUR2B increased the [3H]glibenclamide binding affinity [92]. Neither repaglinide reversibility, nor binding of [3H]repaglinide, were affected by the S1237Y mutation in SUR1 [47, 49].
It is debatable whether rapidly-reversible drugs provide any advantages over slowly-reversible agents for the pancreatic beta cell. Although the trend in therapeutic practice is to treat Type 2 diabetes with "short-acting" KATP channel inhibitors in an attempt to reduce the incidence of hypoglycaemic side effects, drug reversibility in vivo occurs over a very different time scale from that measured in patch clamp experiments, and involves different underlying mechanisms, including drug transport, metabolism and excretion. In clinical practice, even an agent that takes up to an hour to wash off a sulphonylurea receptor is unlikely to cause hypoglycaemia if it is rapidly cleared from the plasma and if dosing is arranged so that the drug levels are sufficiently low between meals. It is of interest, however, that high pre-prandial levels of agents that reverse rapidly at the level of the KATP channel appear to produce fewer hypoglycaemic side effects than slowly-reversible agents. Chlorpropamide, for example, has been found to produce fewer hypoglycaemic side effects than glibenclamide, even though both drugs have prolonged plasma half lives [95, 96, 97]. Also, the production of a modified-release preparation of gliclazide suitable for once-daily administration did not, apparently, increase the risk of hypoglycaemia over the conventional formulation, despite the high levels of drug that remain in the circulation between meals [98]. There could therefore be a case for suggesting that rapidly reversible action at the level of the KATP channel in some way reduces the risk of hypoglycaemic side effects. Whether drug reversibility has any effect on the age-related deterioration of beta cell function or the secondary failure rate of sulphonylurea therapy, are other possibilities that require clarification.
Studies on two new agents, nateglinide and mitiglinide, refute the classical idea that the 'sulphonylurea-binding motif' on SUR1 is selective for sulphonylureas. Nateglinide and mitiglinide, although not containing sulphonylurea groups, exhibit functional properties similar to those of tolbutamide and gliclazide. Thus, they both inhibited Kir6.2/SUR1 with higher affinity than Kir6.2/SUR2 currents, and inhibition of SUR1-type channels was impaired by the S1237Y mutation [48, 49, 50, 97]. Structural comparisons confirm that the nateglinide molecule can be aligned with tolbutamide, a carboxyl group in nateglinide taking the place of the sulphonyl group in tolbutamide, consistent with the idea that these drugs interact with a similar region of SUR1 [49]. Repaglinide, by contrast, is more structurally related to meglitinide, consistent with its similar affinity for SUR1 and SUR2.
Despite the identification of a site involved in sulphonylurea binding, the location of the meglitinide binding site remains elusive. The chimeric approach, used to identify residues involved in sulphonylurea binding, has been of no help because the properties of meglitinide-inhibition of channels containing SUR1 and SUR2 are indistinguishable. Two pieces of evidence, however, point to the involvement of a separate region of the sulphonylurea receptor in the formation of the drug binding pocket. Firstly, low concentrations of [125I]glibenclamide were found to label a peptide fragment corresponding to the N-terminal third of SUR1 [1]. Secondly, expression studies in insect cells showed that proteins corresponding to the N- and C-terminal halves of SUR1 were individually unable to bind [3H]glibenclamide, but formed high-affinity binding sites when expressed together [100]. Further binding experiments in insect cells showed that although TMD0 could be deleted without affecting glibenclamide binding, additional deletion of the cytoplasmic linker (CL3) between TMD0 and TMD1 abolished [3H]glibenclamide binding [101]. Whether CL3 directly contributes to the meglitinide binding region remains, however, to be established.
Kir6.2 could also be labelled by low concentrations of [125I]azidoglibenclamide in the presence of SUR1 [24], suggesting that part of the Kir6.2 subunit might lie close to the sulphonylurea binding site on SUR1. In favour of this idea, some (but not all) studies showed that coexpression with Kir6.2 or Kir6.1 enhanced the [3H]glibenclamide binding affinity of SUR1 or SUR2B-type subunits by five- to ten-fold, indicating either an allosteric or direct effect of Kir6.x on the sulphonylurea binding site [92, 99, 100, 102, 103]. In patch clamp experiments, high-affinity tolbutamide block of Kir6.2/SUR1 currents was abolished by deletion of the N-terminus of Kir6.2, and was impaired by addition of peptides corresponding to the Kir6.2 N-terminus, suggesting that this region of Kir6.2 might be involved in coupling to SUR [104, 105, 106]. This idea is supported by the finding that truncation of the Kir6.2 N-terminus also prevented co-photolabelling of the Kir subunit by [125I]azidoglibenclamide [106].
Interactions between sulphonylureas and nucleotides
The simplistic interpretation of the data discussed above is that the sulphonylureas and glinides are a heterogeneous group of drugs that would exhibit different degrees of cross-reactivity with cardiovascular KATP channels in vivo. Under physiological conditions, however, a number of additional factors need to be taken into consideration, such as the effects of cytoplasmic nucleotides. The finding that sulphonylureas block KATP currents in excised membrane patches by only 50 to 70%, whereas similar drug concentrations almost completely inhibit native KATP channels in beta cells, is a signal that the excised patch does not exactly mimic the physiological situation. The difference is attributable to the presence, in intact cells, of cytoplasmic nucleotides that modulate the degree of sulphonylurea block [87].
Early experiments on native beta cell KATP channels showed that inhibition by tolbutamide and meglitinide was enhanced by MgADP, or by the simultaneous addition of an inhibitory and a stimulatory nucleotide [107, 108]. The explanation for these findings became apparent during experiments on Kir6.2/SUR1 currents in oocytes. 100 µmol/l MgADP causes net activation of Kir6.2/SUR1 currents because the nucleotide simultaneously activates the channels strongly via SUR, and inhibits them weakly via the ATP binding site on Kir6.2. Tolbutamide was found to prevent MgADP activation, resulting in exposure of the inhibitory effect of the nucleotide on Kir6.2 [87]. The overall effect of adding tolbutamide in the presence of MgADP is therefore that channels go from an activated state (net MgADP activation) to a more inhibited state (combined block by ADP and sulphonylurea), and the overall block by the drug appears to be enhanced. Similar findings have been observed using other sulphonylureas and glinides [44, 47, 50, 91, 107, 109].
Whereas cytoplasmic nucleotides enhance the block of SUR1-containing channels by sulphonylureas, however, this is not observed with SUR2-containing channels [44, 47, 109]. KATP channels in isolated cardiac myocytes, for example, have been shown to be relatively unresponsive to glibenclamide during ischaemia [110, 111]. The region responsible for the SUR1/SUR2 difference was localised, by chimera studies, to TMD1 and the first part of NBD1 (Fig. 2) [109]. Exactly how these regions of SUR1 and SUR2 confer differential responsiveness in the combined presence of nucleotides and sulphonylureas remains unclear and is a complicated issue to resolve because of the theoretical potential for TMD1 to interact with other TMDs or NBDs in the same SUR subunit, with other SUR subunits in the octameric complex, or with the Kir6.2 subunits. Whatever the mechanism, the implications are physiologically significant: although drugs like glibenclamide appear to close cardiac-type KATP channels in patch clamp studies carried out without nucleotides, they might be less effective when the channels are opened in vivo by raised ADP concentrations occurring during ischaemia.
Interferences between the binding of nucleotides and sulphonylureas have been detected in [3H]glibenclamide and [32P]azido-ATP binding studies [61, 112, 113, 114, 115, 116]. Glibenclamide was found to impair azido-ATP binding to SUR1 by preventing the stabilisation of nucleotide binding at NBD1 that normally occurs when MgADP or MgATP binds at NBD2 [61]. Such an effect of sulphonylureas on nucleotide binding might underlie the ability of the drugs to prevent MgADP-induced activation of Kir6.2/SUR1 currents. In this respect it would be interesting to know whether sulphonylureas exert similar effects on nucleotide binding to SUR2, but such data is not currently available.
In [3H]glibenclamide binding studies, it was found that Mg-nucleotides decreased the number of high-affinity glibenclamide binding sites by about 75% in membranes expressing SUR2B-type subunits, whereas they marginally reduced the affinity, but not the number, of sulphonylurea binding sites on SUR1 [115, 116]. This led to the speculation that SUR tetramers might undergo a conformational rearrangement in the presence of MgATP and sulphonylurea, resulting, in SUR2B but not SUR1, in the loss of three out of four high-affinity sulphonylurea binding sites [115]. Whether this contributes to the functional differences observed between channels containing SUR1 and SUR2 remains unclear. If it does, the results of the chimera studies [109] would predict that the effect of nucleotides on [3H]glibenclamide binding would depend on the origin of TMD1 and the first part of NBD1.
Interactions between sulphonylureas and KATP channel openers
KATP channel openers such as diazoxide and nicorandil are used in the treatment of hypertension and angina [117]. Opening KATP channels in vascular smooth muscle results in membrane hyperpolarisation (as in pancreatic beta cells), thereby reducing Ca2+ entry and promoting muscle relaxation and vasodilation [9]. The group of KATP channel openers includes both selective and non-selective agents [43]. Nicorandil, pinacidil and cromakalim are relatively selective for SUR2-type compared with SUR1-type KATP channels. Diazoxide, by contrast, activates channels containing either SUR2 or SUR1, and its pancreatic action is utilised therapeutically to reduce insulin secretion in selected subjects with insulinoma or congenital hyperinsulinism [71, 72].
Studies into the molecular basis for KATP channel opener selectivity have identified regions within TMD2 of SUR2 that determine the responsiveness to nicorandil and analogues of cromakalim and pinacidil. Fine mapping of this domain further localised the critical regions to the cytoplasmic linker between TMs13 and 14 (CL7), and residues within the last TM helix—sites that do not exactly overlap with the areas implicated in sulphonylurea binding [43, 51, 52, 57, 118, 119]. The non-identity of sites involved in sulphonylurea and KATP channel opener action is consistent with the results of binding studies on native and recombinant KATP channel subunits, which suggested that the interference between binding of sulphonylureas and the pinacidil analogue, P1075, are governed allosterically, rather than competitively [115, 120].
Studies on native tissues have suggested that diazoxide opens KATP channels in beta cells and smooth muscle, but not those in cardiac muscle [43]. This apparent selectivity for SUR2B over SUR2A is surprising because the SUR2A/B splice variation only modifies the distal C-terminus of NBD2. The explanation could lie in the fact that diazoxide activation is dependent on the presence of Mg-nucleotides [54, 121], the action of which is found to be modified by the C-terminus of SUR2A/B [118, 123]. Addition of MgADP permitted Kir6.2/SUR2A currents to respond to diazoxide [118], and under conditions in which diazoxide activated the channels, its concentration dependence (EC50) was very similar for SUR1 and SUR2 [43]. The location of the diazoxide binding site, like that of the meglitinide binding site, remains unidentified because of the EC50 similarity between the different SUR subunits.
Nicorandil is increasingly used in the treatment of angina, and operates both as a KATP channel opener and a nitric acid donor [117, 124]. Animal studies have implicated roles for both these properties in its therapeutic action. The likelihood that nicorandil will be prescribed for diabetic subjects on sulphonylurea therapy introduces the potential for interactions to occur between the drugs. It is crucially important to understand whether co-administration of nicorandil with a sulphonylurea that can close channels containing SUR2 would result in impaired activity of the opener or reduced inhibitor action. In oocyte experiments, glibenclamide abolished nicorandil-induced activation of Kir6.2/SUR2A and Kir6.2/SUR2B currents, indicating that the effect of the sulphonylurea is dominant over that of the opener [51]. This is perhaps not surprising, as it is well recognised that glibenclamide inhibits native KATP channels in vascular smooth muscle, in an experimental setting that classically involves prior channel activation by cromakalim or diazoxide [9, 125, 126]. Interestingly, although glimepiride abolished the effect of nicorandil on SUR2A-type channels, it was less effective on SUR2B-type channels, consistent with reports that this agent has lower cross-reactivity with vascular KATP channels [125]. As expected, SUR1-selective sulphonylureas, such as gliclazide, did not affect nicorandil activation [51]. Studies are now required to investigate the extent to which the in vitro findings are replicated in a clinical setting. Loss of the KATP channel opening activity but not the nitric oxide donating property of nicorandil would be predicted during concomitant therapy with glibenclamide, and might, in part, explain why nicorandil was found to retain some anti-anginal activity in a small group of glibenclamide-treated patients [127]. A larger controlled study will be required to determine how much of the therapeutic effect of nicorandil is abolished by glibenclamide and other non-selective KATP channel inhibitors.
KATP channel inhibition under physiological conditions
It is clear that the extent of channel inhibition by sulphonylureas and glinides is determined by the presence (or not) of intracellular nucleotides or KATP channel openers, and that channels containing SUR2 subunits exhibit particularly marked differences in the extent of sulphonylurea block under different conditions. In vivo, KATP channels in the cardiovascular system could be modulated by a number of factors in addition to nucleotides, including phophorylation status, phospholipid concentrations and the function of SUR2 accessory subunits such as creatine kinase, adenylate kinase and lactate dehydrogenase [64, 65, 128]. Phosphoinositides, for example, activate Kir6.2/SUR1 and Kir6.2/SUR2A currents, and impair inhibition by sulphonylureas [129, 130, 131]. It remains to be established whether the accessory enzymes associated with SUR2 modify the response to sulphonylureas. In vitro systems are therefore of only limited value when trying to establish whether non-selective KATP channel inhibitors block cardiovascular KATP channels under physiological conditions. In vivo studies might be the most reliable method to detect cardiovascular effects of sulphonylureas and glinides, but must be carried out with therapeutic concentrations of sulphonylureas and without the use of KATP channel openers.
In intact cells, sulphonylureas might also have effects on KATP channels in intracellular organelles. The existence of a mitochondrial KATP channel, for example, has been suggested for many years [132, 133, 134]. In pancreatic beta cells, there is convincing evidence that Kir6.2 and SUR1 subunits are predominantly located in the membranes of secretory granules [135, 136], although their role there remains largely speculative. It is interesting, for example, that SUR1 associates physically with a cAMP-dependent guanine nucleotide exchange factor, cAMP-GEFII (epac2), which is involved in cAMP-stimulated exocytosis (vesicle release) and calcium release from intracellular stores [137, 138, 139]. It has also been found that exocytosis is directly modulated by the ATP/ADP ratio [140], which could theoretically be detected by the granule SUR subunits. The function of the granule KATP channels, the nature of the interaction between cAMP-GEFII and SUR1, and the possibility that they might be modulated by sulphonylureas, will be interesting areas for future work.
Potential consequences of KATP channel blockade in different tissues
The possibility that non-selective KATP channel inhibitors might have extrapancreatic side effects in clinical practice remains highly controversial. Although the UK Prospective Diabetes Study did not demonstrate an increased risk of micro- or macro-vascular events attributable to glibenclamide therapy [141], this study was not designed to detect more subtle, but potentially significant side effects such as modest changes in exercise tolerance. Part of the explanation for the apparent failure to detect side effects could lie in our poor understanding (until recently) of the physiological roles of KATP channels in extra-pancreatic tissues.
Some clarity is now emerging in this area, however, as a result of the phenotypic descriptions of knockout mice lacking different KATP channel subunits (Kir6.2, Kir6.1, SUR1 and SUR2), (Fig. 5). Although mouse physiology is not necessarily equivalent to human physiology, studies on KATP-channel deficient mice might suggest potential consequences of long term non-selective KATP channel inhibition in humans. Whether therapeutic concentrations of agents like glibenclamide could produce such side effects will depend on a number of factors, including the similarities between human (diabetic) and mouse physiology, the efficacy of the drugs under physiological conditions and the consequences of short term or partial channel inhibition compared with a complete channel deficiency extending during development and the entire lifespan of the animal.

Major phenotypes of KATP channel deficient mice. KATP channel deficiency in different organs has been produced in mice by knockout of Kir6.2, Kir6.1, SUR1 or SUR2. The figure shows the SUR subtypes expressed in the affected tissues, and the major phenotypes of the knockout in each tissue
Effects of KATP channel deficiency on glucose homeostasis
The studies on knockout mice showed that glucose homeostasis is regulated by KATP channel activity in a number of tissues, including pancreatic islets, skeletal muscle and the hypothalamus [142, 143, 144, 145, 146, 147, 148, 149]. The best characterised defects in knockout mice are those affecting the function of pancreatic beta cells. Mice lacking either Kir6.2 (Kir6.2−/−) or SUR1 (SUR1−/−) suffered from transient neonatal hypoglycaemia [142, 144], mimicking, on a different time scale, the human condition of congenital hyperinsulinism. Pancreatic beta cells from these animals had no functional KATP channels, were chronically depolarised and had increased basal intracellular calcium concentrations [142, 144]. Acute secretory responses to glucose and sulphonylureas were impaired, consistent with the role of KATP channel closure in triggering first phase insulin release. The rate of insulin secretion declined with age, resulting in normoglycaemia followed, in Kir6.2−/− mice, by hyperglycaemia. The behaviour of Kir6.2−/− mice is therefore similar to that observed in human subjects with the mutation E1506 K in SUR1, which is associated with both congenital hyperinsulism in childhood and diabetes in adults [78, 79]. Morphological studies of pancreas from Kir6.2−/− mice revealed a loss of beta cells and an enhanced rate of apoptosis [143]. Whether this results directly from the chronic depolarisation and increased intracellular Ca2+, or some other mechanism, is unclear. Consistent with the involvement of KATP channels in cAMP-stimulated exocytosis, SUR1−/− mice also displayed impaired insulin release in response to incretins [145, 146].
Kir6.2−/− and SUR2−/−, but not SUR1−/−, mice were more insulin-sensitive than wild-type mice during insulin tolerance tests, reflecting a role of KATP channels in skeletal muscle [142, 144, 147, 148]. Studies on isolated skeletal muscle from Kir6.2−/− and SUR2−/− mice demonstrated increased rates of insulin-stimulated glucose uptake, but the pathway linking KATP channels to the glucose uptake mechanism has not yet been established [147, 148].
Delayed recovery from insulin-induced hypoglycaemia in Kir6.2−/− mice has been attributed to impaired release of glucagon in response to systemic hypoglycaemia or brain glucopaenia, despite the presence of functional pancreatic alpha cells [149]. Central detection of hypoglycaemia is believed to involve glucose-responsive neurones in the ventromedial hypothalamus, which were lacking in Kir6.2−/− mice [149].
KATP channels in the cardiovascular system
KATP channels in cardiac and vascular smooth muscle are believed to be comprised of Kir6.2/SUR2A and Kir6.2/SUR2B subunits, respectively [34, 35]. An additional class of vascular channels with a smaller single channel conductance and lower ATP sensitivity, can be accounted for by the association of the alternative pore subunit, Kir6.1, with SUR2B [25, 27]. Opening KATP channels in vascular smooth muscle causes vasodilation, and occurs physiologically in response to certain neurotransmitters and hypoxia, or pharmacologically during therapy with KATP channel opening drugs [9]. Activation of KATP channels in cardiac muscle, by contrast, is believed to protect the heart during metabolic stress by shortening the cardiac action potential, thereby reducing calcium entry and cardiac work [8]. Cardiac ischaemic preconditioning is a slightly different phenomenon, reflecting the protection against ischaemic damage by prior exposure to hypoxia. It has traditionally been attributed to the opening of KATP channels in mitochondria, resulting in changes in mitochondrial membrane potential and function [134, 150], but the molecular identity of the putative mitochondrial KATP channels has remained surprisingly elusive.
Physiological studies on mice lacking Kir6.2, Kir6.1 or SUR2 are beginning to clarify the roles of cardiac and vascular KATP channels in cardiovascular pathophysiology [151, 152, 153, 154, 155, 156]. Kir6.2 deficient mice had a number of cardiovascular abnormalities during myocardial ischaemia. Although cardiac myocytes from Kir6.2−/− mice did not show the normal action potential shortening during metabolic inhibition, myocardial infarct size was similar to that in wild-type mice following left anterior descending coronary artery (LAD) ligation [151, 152]. LAD occlusion, however, elicited ST-elevation in the ECG of wild type, but not Kir6.2−/−, mice, supporting the idea that KATP channels are involved in the pathogenesis of ischaemic ST-elevation [152, 153]. Surprisingly, ischaemic preconditioning was also absent in Kir6.2−/− mice, implicating a critical involvement of surface membrane (sarcolemmal), rather than mitochondrial, KATP channels in the genesis of this phenomenon in mice [152].
Although a protective role for cardiac KATP channels during hypoxia therefore seems likely, it is less clear that this is their sole function. In this respect, it is interesting that cardiac KATP channels have now been implicated in the normal response to severe stress. Thus, whereas vigorous sympathetic stimulation in wild type mice resulted in adaptive responses in the heart that maintained myocardial function, in Kir6.2−/− mice it caused impaired cardiac performance and calcium handling, resulting in arrhythmias and sudden death [156]. It therefore seems that sympathetic stimulation normally triggers the opening of KATP channels, which enable the heart to maintain an appropriate level of function. Further studies on the knockout mice might lead to the identification of other physiological conditions that similarly require adaptive changes in myocardial KATP channel activity.
Impaired vascular smooth muscle function was a feature of Kir6.1−/− and SUR2−/− mice, manifesting as episodic coronary artery vasospasm and a high rate of sudden death [154, 155]. The pathophysiological picture was similar to the human condition of Prinzmetal angina. Raised systolic and diastolic blood pressure was also observed in SUR2−/−, but not in either Kir6.1−/− or Kir6.2−/−, mice [151, 154, 155]. This suggests that SUR2-containing KATP channels are involved in blood pressure regulation, but that this role cannot be exclusively attributed to channels with either Kir6.1 or Kir6.2 as the pore-forming subunit. Both Kir6.1/SUR2 and Kir6.2/SUR2 channels, or possibly SUR2 coupled to a third type of pore, might be involved.
The mouse studies suggest that adverse cardiovascular effects of non-selective KATP channel inhibition would be largely restricted to periods of ischaemia or severe stress, and might, for example, include impaired ischaemic preconditioning and attenuated electrocardiographic ST changes during myocardial infarction. The idea that sulphonylureas can inhibit cardiac KATP channels despite the high cytoplasmic nucleotide concentrations is supported by the finding that ischaemic preconditioning during coronary angioplasty was prevented by oral administration of 10 mg glibenclamide in human subjects [157]. Glibenclamide also impaired ischaemic ECG changes in dogs [158], leading to the suggestion that the drug might partially mask ST changes in diabetic human subjects with myocardial infarction [159]. The mouse studies support the involvement of KATP channels in ischaemic ST elevation, and should perhaps stimulate a discussion of whether biochemical cardiac markers should be more widely used in the clinical evaluation of diabetic subjects with non-specific symptoms. Despite the potential adverse consequences of cardiac KATP channel inhibition suggested above, however, there is also evidence that blocking sarcolemmal KATP channels might reduce the incidence of reperfusion arrhythmias – an idea that has actually led to the development of a more selective cardiac KATP channel inhibitor [160].
Musculoskeletal system
The role of KATP channels in skeletal muscle is not well understood, although the possibility that they could be involved in glucose uptake has been discussed above. It had been suggested that the opening of KATP channels in skeletal muscle might also underlie the decrease in muscle tension that occurs during fatigue [10]. Physiological studies on muscle function in wild type and Kir6.2−/− mice, however, demonstrated only a small increase in the resting tension in Kir6.2−/− mice following prolonged muscle stimulation, and enhanced age-related deterioration in tetanic force and the rate of recovery after fatigue [161].
Central nervous system
KATP channels are expressed in a number of regions in the brain. In addition to mediating counter-regulatory responses to hypoglycaemia [149], it has been suggested that KATP channel opening during hypoxia might protect neurones by reducing membrane excitability. In support of this idea, Kir6.2−/− mice were more susceptible to hypoxia-induced generalised seizure, perhaps as a result of the loss of KATP channels in the substantia nigra, a region that has been implicated in the control of seizure propagation [162].
Summary and conclusions
KATP channels are located throughout the body in neuroendocrine, muscle and neural tissues, and the list of their extra-pancreatic roles is still growing. The channels in beta cells have a different sulphonylurea receptor subunit from those in cardiac, skeletal and smooth muscle (SUR1 vs SUR2) and, as a result, show different sensitivities to pharmacological agents. The antidiabetic sulphonylureas and glinides fall into two groups, based on their selectivities for channels containing SUR1 or SUR2. Some drugs, such as tolbutamide, chlorpropamide, gliclazide, nateglinide and mitiglinide, seem to be relatively selective for SUR1-type channels, whereas others, such as glibenclamide, glimepiride and repaglinide, inhibit channels containing either SUR1 or SUR2. This is because the latter group can interact with a region of the drug binding site that is common to SUR1 and SUR2.
Adverse consequences of blocking KATP channels containing SUR2 have been notoriously difficult to demonstrate, leading to the continued use of non-selective agents in clinical practice. Studies on KATP channel deficient mice, however, support the idea that the cardiovascular and musculoskeletal consequences of KATP channel inhibition could be subtle, or restricted to rare situations such as myocardial ischaemia or severe stress. A final clinical decision on whether non-selective KATP channel inhibitors are safe to use in the treatment of Type 2 diabetes should depend on the results of studies designed specifically to look for the side effects suggested by the phenotypes of the KATP channel-deficient mice.
Abbreviations
- KATP channel:
-
ATP sensitive potassium channel
- Kir:
-
inwardly-rectifying potassium channel
- SUR:
-
sulphonylurea receptor
- TMD:
-
transmembrane domain
- NBD:
-
nucleotide binding domain
- CL:
-
cytoplasmic linker
- CHI:
-
congenital hyperinsulinism
References
Aguilar-Bryan L, Nichols CG, Wechsler SW et al. (1995) Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268:423–426
Inagaki N, Gonoi T, Clement JP et al. (1995) Reconstitution of IKATP—an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170
Sakura H, Ammala C, Smith PA, Gribble FM, Ashcroft FM (1995) Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta cells, brain, heart and skeletal muscle. FEBS Lett 377:338–344
Ashcroft FM, Gribble FM (1999) ATP-sensitive K+ channels in health and disease. Diabetologia 42:903–919
Aguilar-Bryan L, Bryan J (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev 20:101–135
Seino S, Miki T (2003) Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 81:133–176
Noma A (1983) ATP-regulated K+ channels in cardiac muscle. Nature 305:147–148
Nichols CG, Lederer WJ (1991) Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol 261:H1675–H1686
Quayle JM, Nelson MT, Standen NB (1997) ATP-sensitive and inwardly-rectifying potassium channels in smooth muscle. Physiol Rev 77:1165–1232
Davis NW, Standen NB, Stanfield PR (1991) ATP-dependent potassium channels of muscle cells: their properties, regulation, and possible functions. J Bioenerg Biomembr 23:509–535
Amoroso S, Schmid-Antomarchi H, Fosset M, Lazdunski M (1990) Glucose, sulfonylureas, and neurotransmitter release: role of ATP-sensitive K+ channels. Science 247:852–854
Liss B, Bruns R, Roeper J (1999) Alternative sulfonylurea receptor expression defines metabolic sensitivity of KATP channels in dopaminergic midbrain neurons. EMBO J 18:833–846
Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML (1997) Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390:521–525
Rorsman P (1997) The pancreatic beta cell as a fuel sensor: an electrophysiologist's viewpoint. Diabetologia 40:487–495
Ashcroft FM, Rorsman P (1989) Electrophysiology of the pancreatic beta cell. Prog Biophys Mol Biol 54:87–143
Ashcroft SJ, Weerasinghe LC, Randle PJ (1973) Interrelationship of islet metabolism, adenosine triphosphate content and insulin release. Biochem J 132:223–231
Mertz RJ, Worley JF, Spencer B, Johnson JH, Dukes ID (1996) Activation of stimulus-secretion coupling in pancreatic beta cells by specific products of glucose metabolism. Evidence for privileged signaling by glycolysis. J Biol Chem 271:4838–4845
Eto K, Tsubamoto Y, Terauchi Y et al. (1999) Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283:981–985
Smith PA, Sakura H, Coles B, Gummerson N, Proks P, Ashcroft FM (1997) Electrogenic arginine transport mediates stimulus-secretion coupling in mouse pancreatic beta cells. J Physiol 499:625–635
Reimann F, Gribble FM (2002) Glucose-sensing in glucagon-like peptide-1-secreting cells. Diabetes 51:2757–2763
Gribble FM, Williams L, Simpson AK, Reimann F (2003) A novel glucose-sensing mechanism contributing to Glucagon-Like Peptide-1 secretion from the GLUTag cell line. Diabetes 52:1147–1154
Inagaki N, Tsuura Y, Namba N et al. (1995) Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem 270:5691–5694
Shyng S-L, Nichols CG (1997) Octameric stochiometry of the KATP channel complex. J Gen Physiol 110:655–664
Clement JP, Kunjilwar K, Gonzalez G et al. (1997) Association and stoichiometry of KATP channel subunits. Neuron 18:827–838
Yamada M, Isomoto S, Matsumoto S et al. (1997) Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J Physiol 499:715–720
Thomzig A, Wenzel M, Karschin C et al. (2001) Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane KATP channels. Mol Cell Neurosci 18:671–690
Takano M, Xie LH, Otani H, Horie M (1998) Cytoplasmic terminus domains of Kir6.x confer different nucleotide-dependent gating on the ATP-sensitive K+ channel. J Physiol 512:395–406
Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron 22:537–548
Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387:179–183
Tucker SJ, Gribble FM, Proks P et al. (1998) Molecular determinants of KATP channel inhibition by ATP. EMBO J 17:3290–3296
Lee K, Dixon AK, Richardson PJ, Pinnock RD (1999) Glucose-receptive neurones in the rat ventromedial hypothalamus express KATP channels composed of Kir6.1 and SUR1 subunits. J Physiol 515:439–452
Liu M, Seino S, Kirchgessner AL (1999) Identification and characterization of glucoresponsive neurons in the enteric nervous system. J Neurosci 19:10305–10317
Ibrahim N, Bosch MA, Smart JL et al. (2003) Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology 144:1331–1340
Inagaki N, Gonoi T, Clement JP et al. (1996) A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 16:1011–1017
Isomoto S, Kondo C, Yamada M et al. (1996) A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J Biol Chem 271:24321–24324
Chutkow WA, Makielski JC, Nelson DJ, Burant CF, Fan Z (1999) Alternative splicing of sur2 Exon 17 regulates nucleotide sensitivity of the ATP-sensitive potassium channel. J Biol Chem 274:13656–13665
Chutkow WA, Simon MC, Le Beau MM, Burant CF (1996) Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes 45:1439–1445
Gros L, Trapp S, Dabrowski M, Ashcroft FM, Bataille D, Blache P (2002) Characterization of two novel forms of the rat sulphonylurea receptor SUR1A2 and SUR1BDelta31. Br J Pharmacol 137:98–106
Hambrock A, Preisig-Muller R, Russ U et al. (2002) Four novel splice variants of sulfonylurea receptor 1. Am J Physiol Cell Physiol 283: C587-C598
Sakura H, Trapp S, Liss B, Ashcroft FM (1999) Altered functional properties of KATP channel conferred by a novel splice variant of SUR1. J Physiol 521:337–350
Tusnady GE, Bakos E, Varadi A, Sarkadi B (1997) Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Lett 402:1–3
Conti LR, Radeke CM, Shyng SL, Vandenberg CA (2001) Transmembrane topology of the sulfonylurea receptor SUR1. J Biol Chem 276:41270–41278
Ashcroft FM, Gribble FM (2000) New windows on the mechanism of action of KATP channel openers. Trends Pharmacol Sci 21:439–445
Gribble FM, Tucker SJ, Seino S, Ashcroft FM (1998) Tissue specificity of sulphonylureas: studies on cloned cardiac and beta cell KATP channels. Diabetes 47:1412–1418
Gribble FM, Ashcroft FM (1999) Differential sensitivity of β-cell and extrapancreatic KATP channels to gliclazide. Diabetologia 42:845–848
Song DK, Ashcroft FM (2001) Glimepiride block of cloned β-cell, cardiac and smooth muscle K-ATP channels. Brit J Pharmacol 133:193–199
Dabrowski M, Wahl P, Holmes WE, Ashcroft FM (2001) Effect of repaglinide on cloned beta cell, cardiac and smooth muscle types of ATP-sensitive potassium channel. Diabetologia 44:747–756
Reimann F, Proks P, Ashcroft FM (2001) Effects of mitiglinide (S 21403) on Kir6.2/SUR1, Kir6.2/SUR2A and Kir6.2/SUR2B types of ATP-sensitive potassium channel. Br J Pharmacol 132:1542–1548
Hansen AM, Christensen IT, Hansen JB, Carr RD, Ashcroft FM, Wahl P (2002) Differential interactions of nateglinide and repaglinide on the human beta cell sulphonylurea receptor 1. Diabetes 51:2789–95
Chachin M, Yamada M, Fujita A, Matsuoka T, Matsushita K, Kurachi Y (2003) Nateglinide, a D-phenylalanine derivative lacking either a sulfonylurea or benzamido moiety, specifically inhibits pancreatic beta cell-type KATP channels. J Pharmacol Exp Ther 304:1025–1032
Reimann F, Ashcroft FM, Gribble FM (2001) Structural basis for the interference between nicorandil and sulfonylurea action. Diabetes 50:2253–2259
Uhde I, Toman A, Gross I, Schwanstecher C, Schwanstecher M (1999) Identification of the potassium channel opener site on sulfonylurea receptors. J Biol Chem 274:28079–28082
Gribble FM, Tucker SJ, Haug T, Ashcroft FM (1998) MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci USA 95:7185–7190
Gribble FM, Tucker SJ, Ashcroft FM (1997) The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J 16:1145–1152
Shyng S, Ferrigni T, Nichols CG (1997) Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol 110:643–654
Nichols CG, Shyng SL, Nestorowicz A et al. (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272:1785–1787
Babenko AP, Gonzalez G, Bryan J (2000) Pharmaco-topology of sulfonylurea receptors. Separate domains of the regulatory subunits of KATP channel isoforms are required for selective interaction with K+ channel openers. J Biol Chem 275:717–720
Senior AE, al-Shawi MK, Urbatsch IL (1995) The catalytic cycle of P-glycoprotein. FEBS Lett 377:285–289
Higgins CF (1992) ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8:67–113
Ueda K, Inagaki N, Seino S (1997) MgADP antagonism to Mg2+-independent ATP binding of the sulfonylurea receptor SUR1. J Biol Chem 272:22983–22986
Ueda K, Komine J, Matsuo M, Seino S, Amachi T (1999) Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc Natl Acad Sci USA 96:1268–1272
Zingman LV, Alekseev AE, Bienengraeber M et al. (2001) Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31:233–245
Matsuo M, Dabrowski M, Ueda K, Ashcroft FM (2002) Mutations in the linker domain of NBD2 of SUR inhibit transduction but not nucleotide binding. EMBO J 21:4250–4258
Carrasco AJ, Dzeja PP, Alekseev AE et al. (2001) Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA 98:7623–7628
Crawford RM, Ranki HJ, Botting CH, Budas GR, Jovanovic A (2002) Creatine kinase is physically associated with the cardiac ATP-sensitive K+ channel in vivo. FASEB J 16:102–104
Matsuo M, Kioka N, Amachi T, Ueda K (1999) ATP binding properties of the nucleotide-binding folds of SUR1. J Biol Chem 274:37479–37482
Bienengraeber M, Alekseev AE, Abraham MR et al. (2000) ATPase activity of the sulfonylurea receptor: a catalytic function for the KATP channel complex. FASEB J 14:1943–1952
Matsuo M, Tanabe K, Kioka N, Amachi T, Ueda K (2000) Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J Biol Chem 275:28757–28763
Thomas PM, Cote GJ, Wohllk N et al. (1995) Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268:426–429
Kane C, Shepherd RM, Squires PE et al. (1996) Loss of functional KATP channels in pancreatic β-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nature Med 2:1344–1347
Huopio H, Shyng SL, Otonkoski T, Nichols CG (2002) KATP channels and insulin secretion disorders. Am J Physiol 283:E207–E216
Glaser B, Thornton P, Otonkoski T, Junien C (2000) Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed 82:F79–F86
Lonlay P de, Fournet JC, Touati G et al. (2002) Heterogeneity of persistent hyperinsulinaemic hypoglycaemia. A series of 175 cases. Eur J Pediatr 161:37–48
Cartier EA, Conti LR, Vandenberg CA, Shyng SL (2001) Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci 98:2882–2887
Tanizawa Y, Matsuda K, Matsuo M et al. (2000) Genetic analysis of Japanese patients with persistent hyperinsulinemic hypoglycemia of infancy: nucleotide-binding fold-2 mutation impairs cooperative binding of adenine nucleotides to sulfonylurea receptor 1. Diabetes 49:114–120
Shyng SL, Ferrigni T, Shepard JB et al. (1998) Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes 47:1145–1151
Reimann F, Huopio H, Dabrowski M et al. (2003) Characterisation of new KATP-channel mutations associated with congenital hyperinsulinism in the Finnish population Diabetologia 46:241–249
Huopio H, Reimann F, Ashfield R et al. (2000) Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest 106:897–906
Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M (2003) A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 361:301–307
Hani EH, Boutin P, Durand E et al. (1998) Missense mutations in the pancreatic islet beta cell inwardly rectifying K+ channel gene (KIR6.2/BIR): a meta-analysis suggests a role in the polygenic basis of Type II diabetes mellitus in Caucasians. Diabetologia 41:1511–1515
Gloyn AL, Weedon MN, Owen KR et al. (2003) Large-Scale Association Studies of Variants in Genes Encoding the Pancreatic beta cell KATP Channel Subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) Confirm That the KCNJ11 E23K Variant Is Associated With Type 2 Diabetes. Diabetes 52:568–572
Love-Gregory L, Wasson J, Lin J, Skolnick G, Suarez B, Permutt MA (2003) An E23K single nucleotide polymorphism in the islet ATP-sensitive potassium channel gene (Kir6.2) contributes as much to the risk of Type II diabetes in Caucasians as the PPAR Pro12Ala variant. Diabetologia 46:136–137
Schwanstecher C, Neugebauer B, Schulz M, Schwanstecher M (2002) The common single nucleotide polymorphism E23K in Kir6.2 sensitizes pancreatic beta cell ATP-sensitive potassium channels toward activation through nucleoside diphosphates. Diabetes 51 (Suppl 3):S363–S367
Schwanstecher C, Meyer U, Schwanstecher M (2002) Kir6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic beta cell ATP-sensitive K+ channels. Diabetes 51:875–879
Hart LM 't, Haeften TW van, Dekker JM, Bot M, Heine RJ, Maassen JA (2002) Variations in insulin secretion in carriers of the E23K variant in the KIR6.2 subunit of the ATP-sensitive K+ channel in the beta cell. Diabetes 51:3135–3138
Nielsen EM, Hansen L, Carstensen B et al. (2003) The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of Type 2 diabetes. Diabetes 52:573–577
Gribble FM, Tucker SJ, Ashcroft FM (1997) The interaction of nucleotides with the tolbutamide block of K-ATP currents: a reinterpretation. J Physiol 504:35–45
Gros L, Virsolvy A, Salazar G, Bataille D, Blache P (1999) Characterization of low-affinity binding sites for glibenclamide on the Kir6.2 subunit of the beta cell KATP channel. Biochem Biophys Res Commun 257:766–770
Proks P, Reimann F, Green N, Gribble FM, Ashcroft FM (2002) Sulphonylurea stimulation of insulin secretion. Diabetes 51 [Suppl 3]:S368–S376
Schmid-Antomarchi H, De Weille J, Fosset M, Lazdunski M (1987) The receptor for antidiabetic sulfonylureas controls the activity of the ATP-modulated K+ channel in insulin-secreting cells. J Biol Chem 262:15840–15844
Ashfield R, Gribble FM, Ashcroft SJH, Ashcroft FM (1999) Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes 48:1341–1347
Hambrock A, Löffler-Walz C, Russ U, Lange U, Quast U (2001) Characterization of a mutant sulfonylurea receptor SUR2B with high affinity for sulfonylureas and openers: differences in the coupling to Kir6.x subtypes. Mol Pharmacol 60:190–199
Muller G, Hartz D, Punter J, Okonomopulos R, Kramer W (1994) Differential interaction of glimepiride and glibenclamide with the beta cell sulfonylurea receptor. I. Binding characteristics. Biochim Biophys Acta 1191:267–277
Hu S, Wang S, Fanelli B et al. (2000) Pancreatic beta cell KATP channel activity and membrane-binding studies with nateglinide: a comparison with sulfonylureas and repaglinide. J Pharmacol Exp Ther 293:444–452
UK Prospective Diabetes Study Group (1995) United Kingdom Prospective Diabetes Study (UKPDS). 13: Relative efficacy of randomly allocated diet, sulphonylurea, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 310:83–88
Clarke BF, Campbell IW (1975) Long-term comparative trial of glibenclamide and chlorpropamide in diet-failed, maturity-onset diabetics. Lancet 1:246–248
Jennings AM, Wilson RM, Ward JD (1989) Symptomatic hypoglycemia in NIDDM patients treated with oral hypoglycemic agents. Diabetes Care 12:203–208
Drouin P (2000) Diamicron MR once daily is effective and well tolerated in type 2 diabetes: a double-blind, randomized, multinational study. J Diabetes Complications 14:185–191
Sunaga Y, Gonoi T, Shibasaki T et al. (2001) The effects of mitiglinide (KAD-1229), a new anti-diabetic drug, on ATP-sensitive K+ channels and insulin secretion: comparison with the sulfonylureas and nateglinide. Eur J Pharmacol 431:119–125
Mikhailov MV, Mikhailova EA, Ashcroft SJ (2000) Investigation of the molecular assembly of beta cell KATP channels. FEBS Lett 482:59–64
Mikhailov MV, Mikhailova EA, Ashcroft SJ (2001) Molecular structure of the glibenclamide binding site of the beta cell KATP channel. FEBS Lett 499:154–160
Russ U, Hambrock A, Artunc F et al. (1999) Coexpression with the inward rectifier K+ channel Kir6.1 increases the affinity of the vascular sulfonylurea receptor SUR2B for glibenclamide. Mol Pharmacol 56:955–961
Dorschner H, Brekardin E, Uhde I, Schwanstecher C, Schwanstecher M (1999) Stoichiometry of sulfonylurea-induced ATP-sensitive potassium channel closure. Mol Pharmacol 55:1060–1066
Reimann F, Tucker SJ, Proks P, Ashcroft FM (1999) Involvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol 518:325–336
Koster JC, Sha Q, Nichols CG (1999) Sulfonylurea and K+-channel opener sensitivity of KATP channels. Functional coupling of Kir6.2 and SUR1 subunits. J Gen Physiol 114:203–213
Babenko AP, Bryan J (2002) SUR-dependent modulation of KATP channels by an N-terminal Kir6.2 peptide. Defining intersubunit gating interactions. J Biol Chem 277:43997–44004
Zunkler BJ, Lins S, Ohno-Shosaku T, Trube G, Panten U (1988) Cytosolic ADP enhances the sensitivity to tolbutamide of ATP-dependent K+ channels from pancreatic B-cells. FEBS Lett 239:241–244
Schwanstecher C, Dickel C, Panten U (1992) Cytosolic nucleotides enhance the tolbutamide sensitivity of the ATP-dependent K+ channel in mouse pancreatic B cells by their combined actions at inhibitory and stimulatory receptors. Mol Pharmacol 41:480–486
Reimann F, Dabrowski M, Jones P, Gribble F, Ashcroft FM (2003) Analysis of the differential modulation of sulphonylurea block of β-cell and cardiac KATP channels by Mg-nucleotides. J Physiol 547:159–168
Venkatesh N, Lamp ST, Weiss JN (1991) Sulfonylureas, ATP-sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle. Circ Res 69:623–637
Findlay I (1993) Sulphonylurea drugs no longer inhibit ATP-sensitive K+ channels during metabolic stress in cardiac muscle. J Pharmacol Exp Ther 266:456–467
Niki I, Nicks JL, Ashcroft SJ (1990). The beta cell glibenclamide receptor is an ADP-binding protein. Biochem J 268:713–718
Schwanstecher M, Loser S, Brandt C, Scheffer K, Rosenberger F, Panten U (1992) Adenine nucleotide-induced inhibition of binding of sulphonylureas to their receptor in pancreatic islets. Br J Pharmacol 105:531–534
Hambrock A, Loffler-Walz C, Kurachi Y, Quast U (1998) Mg2+ and ATP dependence of KATP channel modulator binding to the recombinant sulphonylurea receptor, SUR2B. Br J Pharmacol 125:577–583
Loffler-Walz C, Hambrock A, Quast U (2002) Interaction of KATP channel modulators with sulfonylurea receptor SUR2B: implication for tetramer formation and allosteric coupling of subunits. Mol Pharmacol 61:407–614
Hambrock A, Loffler-Walz C, Quast U (2002) Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br J Pharmacol 136:995–1004
Patel DJ, Purcell HJ, Fox KM (1999) Cardioprotection by opening of the KATP channel in unstable angina. Is this a clinical manifestation of myocardial preconditioning? Results of a randomized study with nicorandil. CESAR 2 investigation. Clinical European studies in angina and revascularization. Eur Heart J 20:51–57
D'hahan N, Moreau C, Prost AL et al. (1999) Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci USA 96:12162–12167
Moreau C, Jacquet H, Prost AL, D'hahan N, Vivaudou M (2000) The molecular basis of the specificity of action of KATP channel openers. EMBO J 19:6644–6651
Bray KM, Quast U (1992) A specific binding site for K+ channel openers in rat aorta. J Biol Chem 267:11689–11692
Terzic A, Jahangir A, Kurachi Y (1995). Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+ channel-opening drugs. Am J Physiol 269:C525–C545
Reimann F, Gribble FM, Ashcroft FM (2000) Differential response of KATP channels containing SUR2A or SUR2B subunits to nucleotides and pinacidil. Mol Pharmacol 58:1318–1325
Matsuoka T, Matsushita K, Katayama Y et al. (2000) C-terminal tails of sulfonylurea receptors control ADP-induced activation and diazoxide modulation of ATP-sensitive K+ channels. Circ Res 87:873–880
Cogolludo AL, Perez-Vizcaino F, Fajardo S, Ibarra M, Tamargo J (1999) Effects of nicorandil as compared to mixtures of sodium nitroprusside and levcromakalim in isolated rat aorta. Br J Pharmacol 126:1025–1033
Bijlstra PJ, Lutterman JA, Russel FGM, Thien T, Smits P (1996) Interaction of sulphonylurea derivatives with vascular ATP-sensitive potassium channels in humans. Diabetologia 39:1083–1090
Lawrence CL, Proks P, Rodrigo GC et al. (2001) Gliclazide produces high-affinity block of KATP channels in mouse isolated pancreatic β-cells but not rat heart or arterial smooth muscle cells. Diabetologia 44:1019–1025
Hata N, Takano M, Kunimi T, Kishida H, Takano T (2001) Lack of antagonism between nicorandil and sulfonylurea in stable angina pectoris. Int J Clin Pharmacol Res 21:59–63
Carrasco AJ, Dzeja PP, Alekseev AE et al. (2002) M-LDH serves as a sarcolemmal KATP channel subunit essential for cell protection against ischemia. EMBO J 21:3936–3948
Fan Z, Makielski JC (1999) Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J Gen Physiol 114:251–269.
Shyng SL, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282:1138–1141
Krauter T, Ruppersberg JP, Baukrowitz T (2001) Phospholipids as modulators of KATP channels: distinct mechanisms for control of sensitivity to sulphonylureas, K+ channel openers, and ATP. Mol Pharmacol 59:1086–1093
Inoue I, Nagase H, Kishi K, Higuti T (1991) ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 352:244–247
Garlid KD (1996) Cation transport in mitochondria—the potassium cycle. Biochim Biophys Acta 1275:123–126
Grover GJ, Garlid KD (2000) ATP-sensitive potassium channels: a review of their cardioprotective pharmacology. J Mol Cell Cardiol 32:677–695
Ozanne SE, Guest PC, Hutton JC, Hales CN (1995) Intracellular localization and molecular heterogeneity of the sulphonylurea receptor in insulin-secreting cells. Diabetologia 38:277–282
Geng X, Li L, Watkins S, Robbins PD, Drain P (2003) The insulin secretory granule is the major site of KATP channels of the endocrine pancreas. Diabetes 52:767–776
Ozaki N, Shibasaki T, Kashima Y et al. (2000) cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2:805–811
Kashima Y, Miki T, Shibasaki T et al. (2001) Critical role of cAMP-GEFII–Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem 276:46046–46053
Kang G, Chepurny OG, Holz GG (2001) cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta cells. J Physiol 536:375–385
Barg S, Eliasson L, Renstrom E, Rorsman P (2002) A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta cells. Diabetes 51 Suppl 1:S74–S82
UK Prospective Diabetes Study Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Miki T, Nagashima K, Tashiro F et al. (1998) Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 95:10402–10406.
Miki T, Iwanaga T, Nagashima K, Ihara Y, Seino S (2001) Roles of ATP-sensitive K+ channels in cell survival and differentiation in the endocrine pancreas. Diabetes 50 (Suppl 1):S48–S51
Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J (2000) Sur1 knockout mice. A model for KATP channel-independent regulation of insulin secretion. J Biol Chem 275:9270–9277
Nakazaki M, Crane A, Hu M et al. (2002) cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes 51:3440–3449
Shiota C, Larsson O, Shelton KD et al. (2002) Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem 277:37176–37183
Chutkow WA, Samuel V, Hansen PA et al. (2001) Disruption of Sur2-containing KATP channels enhances insulin-stimulated glucose uptake in skeletal muscle. Proc Natl Acad Sci USA 98:11760–11764
Miki T, Minami K, Zhang L et al. (2002) ATP-sensitive potassium channels participate in glucose uptake in skeletal muscle and adipose tissue. Am J Physiol 283:E1178–E1184
Miki T, Liss B, Minami K et al. (2001) ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci 4:507–512
Gross GJ, Fryer RM (1999) Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res 84:973–979
Suzuki M, Li RA, Miki T et al. (2001) Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ Res 88:570–577
Suzuki M, Sasaki N, Miki T et al. (2002) Role of sarcolemmal KATP channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest 109:509–516
Li RA, Leppo M, Miki T, Seino S, Marban E (2000) Molecular basis of electrocardiographic ST-segment elevation. Circ Res 87:837–839
Miki T, Suzuki M, Shibasaki T et al. (2002) Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med 8:466–472
Chutkow WA, Pu J, Wheeler MT et al. (2002) Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 KATP channels. J Clin Invest 110:203–208
Zingman LV, Hodgson DM, Bast PH et al. (2002) Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA 99:13278–13283
Tomai F, Crea F, Gaspardone A et al. (1994) Ischemic preconditioning during coronary angioplasty is prevented by glibenclamide, a selective ATP-sensitive K+ channel blocker. Circulation 90:700–705
Kondo T, Kubota I, Tachibana H, Yamaki M, Tomoike H (1996) Glibenclamide attenuates peaked T wave in early phase of myocardial ischemia. Cardiovasc Res 31:683–687
Wilde AA (1996) ATP-sensitive potassium channels, transmural ischemia and the ECG implications for the non-insulin dependent diabetic patient? Cardiovasc Res 31:688–690
Dhein S, Pejman P, Krusemann K (2000) Effects of the IK.ATP blockers glibenclamide and HMR1883 on cardiac electrophysiology during ischemia and reperfusion. Eur J Pharmacol 398:273–284
Gong B, Miki T, Seino S, Renaud JM (2000) A KATP channel deficiency affects resting tension, not contractile force, during fatigue in skeletal muscle. Am J Physiol 279:C1351–C1358
Yamada K, Ji JJ, Yuan H et al. (2001) Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science 292:1543–1546
Chang G, Roth CB (2001) Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 293:1793–1800
Acknowledgements
We thank the Wellcome Trust and Diabetes UK for their support. F.M.G. is a Wellcome Trust Clinician Scientist Fellow and F.R. is a Diabetes UK RD Lawrence Fellow. We also thank Prof. F.M. Ashcroft (Oxford, UK) for her friendship and for giving us the opportunity to work in the KATP channel field, and Dr. M. Evans (Cambridge, UK) for helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gribble, F.M., Reimann, F. Sulphonylurea action revisited: the post-cloning era. Diabetologia 46, 875–891 (2003). https://doi.org/10.1007/s00125-003-1143-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1143-3