


Abstract
Acinetobacter baumannii has become a pathogen of increasing medical importance because of the emergence of multidrug-resistant strains and the high rate of mortality of infected patients. Promising animal study results have been reported recently with active and passive immunization against A. baumannii virulence factors. In the present study, a monoclonal IgG3 antibody, 8E3, was developed with specificity for the K2 capsular polysaccharide of A. baumannii, and its therapeutic potential was assessed. 8E3 enhanced macrophage-mediated bactericidal activity against the A. baumannii clinical strain AB899. However, 8E3 treatment (passive immunization) of AB899-infected mice led to a substantial increase in mortality and to substantial increases in bacterial load in blood, lung, and in splenic samples. In vitro investigations showed a large binding capacity in the supernatant of bacterial cultures, suggesting that shed capsule components act as a binding sink for 8E3. Investigations of 8E3 pharmacokinetics in mice demonstrated that unbound concentrations of the antibody dropped below detection limits within 24 hours after a 200 mg/kg dose. However, total concentrations of antibody declined slowly, with an apparent terminal half-life (t1/2) of 6.7–8.0 days, suggesting that the vast majority of 8E3 in blood is bound (e.g., with soluble capsule components in blood). We hypothesize that high concentrations of 8E3-capsule immune complexes act to inhibit bacterial clearance in vivo. To the best of our knowledge, this is the first demonstration of antibody-dependent enhancement of A. baumannii infection, and these observations highlight the complexity of antibody-based therapy for A. baumannii infections.
Introduction
Immunoglobulin plays an important role in humoral immunity, as it is a key factor in the activation of effector functions that act to eliminate invading microorganisms. However, evidence is accumulating that, in some circumstances, antibody can augment infection. Antibody-dependent enhancement (ADE) of infection was first reported by Hawkes (1964) in the context of Murray Valley viral encephalitis. Although evidence of ADE of infection in human subjects has only been reported for Dengue virus (Vaughn et al., 2000), this phenomenon has been demonstrated for other viruses in animal infection models, including infections caused by Dengue virus, human immunodeficiency virus, Ebola virus, and Zika virus (Halstead, 1988; Tóth et al., 1994; Takada et al., 2003; Sasaki et al., 2013; Bardina et al., 2017). ADE of viral infection typically proceeds through the interaction of complexes of antibody-virus or complement-virus with Fc receptors or complement receptors, leading to suppressed innate and adaptive immunity, enhanced cellular adherence and entry, and increased intracellular replication of the virus (Takada and Kawaoka, 2003; Tirado and Yoon, 2003; Halstead et al., 2010; Ubol and Halstead, 2010). Few demonstrations of antibody-mediated enhancement of bacterial infection have been reported. Weiser et al. (2003) demonstrated IgA1-dependent enhancement of pneumococcal adherence to epithelial cells, where Fab fragments of anti-capsule IgA1 (generated by IgA1 protease cleavage) exposed phosphorylcholine masked by capsular polysaccharide of Streptococcus pneumoniae, thereby augmenting bacterial binding to epithelial cells (via cross-linking with the phosphorylcholine receptor). Antibodies directed against Mycobacterium tuberculosis were also shown to promote the invasion of M. tuberculosis to A549 human lung epithelial cells (Zimmermann et al., 2016). HA-1A, a human IgM monoclonal antibody (mAb) directed against lipid A, has been shown to enhance mortality in a canine model of Gram-negative sepsis, and antibodies targeting the protective antigen of Bacillus anthracis were also found to enhance the lethal toxin activity in rats, but the corresponding mechanisms were not elucidated (Quezado et al., 1993; Little et al., 2011).
Acinetobacter baumannii has recently been identified as one of the three most troublesome pathogens to human health by the World Health Organization because of rapid emergence of antimicrobial-resistant strains (Perez et al., 2007, 2008; Doi et al., 2009; Higgins et al., 2010). Infections with A. baumannii include ventilator-associated pneumonia, bacteremia, skin and wound infections, urinary tract infections, and meningitis-ventriculitis as a consequence of neurosurgical interventions (Michalopoulos and Falagas, 2010). The overall mortality rates associated with A. baumannii infection range from 19% to 54% (Gaynes et al., 2005). The increasing prevalence of extensively drug-resistant (XDR) (i.e., resistant to all antibiotics except for polymyxins or tigecycline) and pan-drug–resistant (i.e., resistant to all approved antibiotics) A. baumannii strains is particularly concerning (Howard et al., 2012; Magiorakos et al., 2012; Shlaes et al., 2013). In view of the emerging treatment of A. baumannii infection, there has been increased consideration of vaccination and antibody-based therapy as potential therapeutic options. Active and passive immunization against various virulence factors, including outer membrane protein A, outer membrane protein 22, outer membrane vesicles, and capsular polysaccharides, have been shown to confer protection against multidrug-resistant or XDR A. baumannii clinical isolates in animal infection models (McConnell et al., 2011; Luo et al., 2012; Russo et al., 2013; Huang et al., 2016; Zhang et al., 2016).
In this report, the therapeutic potential of an mAb, 8E3, directed against the K2 capsular polysaccharide of A. baumannii was assessed. Surprisingly, 8E3 treatment enhanced A. baumannii infection in a mouse pneumonia model. Studies designed to generate insights into the potential mechanisms for the observed ADE and studies of 8E3 pharmacokinetics are presented. The reported results emphasize that the therapeutic utility of mAbs in the management of XDR bacterial infections may be more complicated than anticipated.
Materials and Methods
Bacterial Strains and Cell Culture
Clinical isolates of A. baumannii 854, 899, 955, and 985 were obtained from Walter Reed Medical Center (Bethesda, MD) and were provided by D. Craft; HUMC1 and HUMC6 were isolated from Harbor-UCLA Medical Center (Los Angeles, CA) and was given by B. Spellberg (Luo et al., 2012); AB307-0294 and “Kelly lung” were isolated from patients hospitalized at Erie County Medical Center (Buffalo, NY) (Russo et al., 2009); and AB8407 was obtained from University Hospitals of Cleveland (Cleveland, OH). Capsule types in A. baumannii (AB) have not been completely defined (Kenyon and Hall, 2013), and the capsule types of AB307-0294, AB854/AB899/HUMC6/“Kelly lung”/AB8407, HUMC1, AB985, and AB955 were designated as K1, K2, K4, K11, and K16, respectively, based on wzc classification developed in the laboratory of T. Russo (unpublished data). All strains were grown in lysogeny broth (LB) and maintained at −80°C in 50% LB medium and 50% glycerol. Murine macrophages J774A.1 [TIB-67; American Type Culture Collection (ATCC), Manassas, VA] and RAW 264.7 (TIB-71; ATCC) were grown in Dulbecco’s modified Eagle’s medium (30-2002; ATCC) supplemented with 10% heat-inactivated fetal bovine serum. Human lung epithelial cells NCI-H292 [H292] (CRL-1848; ATCC) were cultured in RPMI 1640 (30-2001; ATCC) with 10% fetal bovine serum for bacterial adherence assay.
Development of Anti-K2 Capsule mAb 8E3
The mAb 8E3 that binds specifically to the capsule of A. baumannii K2 capsule type was developed using a method described previously (Kennett, 1979; Haase et al., 1991). In brief, the capsular polysaccharide was extracted from A. baumannii 854 using the method described by Pelkonen et al. (1988). Female BALB/c mice were injected subcutaneously with 50 µg of K2 capsules in 100 µl of normal saline every 4 weeks. Four days after the second immunization, mouse splenocytes were isolated and fused with SP2/0-Ag14 (CRL-1581; ATCC) cells to obtain hybridomas. Binding of antibodies to the K2 capsule was initially assessed by screening hybridoma supernatants using immunodot and Western blot assays. Hybridomas that produced antibodies that reacted with the K2 capsule were cloned by limiting dilutions and grown in serum-free medium (Thermo Fisher Scientific, Waltham, MA). Monoclonal antibodies were subsequently purified using a protein G affinity column (GE Healthcare Life Sciences, Marlborough, MA). The isotype of the final selected mAb 8E3 was determined to be IgG3 (κ) using Pierce Rapid Isotyping Kits (Thermo Fisher Scientific).
Generation of Antibody F(ab′)2 Fragments
The F(ab′)2 fragment of 8E3 was produced via pepsin digestion (Thermo Fisher Scientific) as described by the manufacturer. Ten milligrams of 8E3 was incubated with 250 µl of immobilized pepsin slurry in 1.5 ml of 20 mM sodium acetate (pH 4.5) at 37°C for 60 minutes with rapid agitation. Pepsin was inactivated by 1.5 ml of 10 mM Tris-HCl (pH 7.5) at the end of the reaction, and crude digest was collected via centrifugation at 1000g for 5 minutes. To obtain pure F(ab′)2, small Fc fragments and undigested IgG were removed using dialysis (50,000 molecular weight cutoff) and a protein A column (Thermo Fisher Scientific), respectively. The purity of the generated F(ab′)2 was confirmed using 4%–15% SDS-PAGE (Bio-Rad, Hercules, CA).
Detection of Antibody Binding to AB899 by Flow Cytometry
Binding of 8E3 to live AB899 was assessed by flow cytometry as described previously (Russo et al., 2013). Briefly, 1 × 106 colony-forming units (CFU) of AB899 were incubated with 8E3 at 1, 5, 10, 100, and 200 nM in 100 µl of phosphate-buffered saline (PBS) at 37°C for 60 minutes. Samples were then washed with 1 ml of PBS. Binding of 8E3 to bacteria was detected using 200× diluted goat anti-mouse IgG (Fc-specific)-R-phycoerythrin conjugates (Thermo Fisher Scientific) via incubation at 37°C for 30 minutes. Samples were washed and resuspended in 0.5 ml of PBS for assessment of binding using flow cytometry. A mouse IgG3 (mIgG3) that binds keyhole limpet hemocyanin (Novus Biologicals, Centennial, CO) was used as an isotype control. To assess the binding of F(ab′)2 of 8E3 to AB899, 100× diluted goat anti-mouse IgG (H+L)-fluorescein isothiocyanate (FITC) conjugate (Thermo Fisher Scientific) was used as the detection antibody, and F(ab′)2 fragments of the mIgG3 isotype were generated for use as a negative control.
Opsonophagocytosis Assay
A antibody-dependent phagocytosis assay was performed based on a modification of a previously used method (Mosser and Zhang, 2008; Russo et al., 2013). Macrophages J774A.1 and RAW 264.7 were primed with 100 U/ml mouse interferon γ (R&D Systems, Minneapolis, MN) for 6 hours and then activated with 10 ng/ml lipopolysaccharide (InvivoGen, San Diego, CA) overnight at 37°C. The clinical isolate AB899 was preopsonized with 8E3 at a concentration range of 6.67–6667 nM (i.e., 1–1000 µg/ml) for 60 minutes at room temperature. The opsonized bacteria were then exposed to the activated murine macrophages (5 × 105/well) in 24-well plates to give a multiplicity of infection (MOI) of 1:6 to 312:1, bacteria to macrophages. Plates were centrifuged at 250g for 10 minutes to allow bacteria adhere to the monolayer of macrophages, and incubated at 37°C for 60 minutes. Macrophages were lysed with 0.5 mg/ml saponin to release live intracellular bacteria at the end of the experiment, and total bacteria in each well (i.e., bacteria in supernatant, bacteria bound to macrophage surface, and released intracellular bacteria) were enumerated by serial 10-fold dilutions and plating on LB agar plates. Results were expressed as the percentage of survival of bacteria compared with the CFU of mIgG3 isotype control.
Enzyme-Linked Immunosorbent Assay Quantification of mAb 8E3
Enzyme-linked immunosorbent assay (ELISA) methods were developed to measure 8E3 concentration in LB medium and in mouse plasma. The K2 capsular polysaccharide of AB899 was extracted using the method described by Pelkonen et al. (1988), and isolated capsules were dried and weighed. Samples of 8E3 were prepared at a range of concentrations (0, 50, 100, 200, 400, 600, 800, and 1000 ng/ml) in PBS (pH 7.4) with 1% (v/v) LB medium or male BALB/c mouse plasma. Standard curves were fitted with quadratic equations with coefficients of determination (R2) greater than 0.99. The working range of the assay was 50–1000 ng/ml, with a lower limit of quantification (LLOQ) of 50 ng/ml and a lower limit of detection (LLOD) of 30 ng/ml, which correspond to an LLOQ of 5 µg/ml and an LLOD of 3 µg/ml in undiluted plasma or bacterial culture samples.
Assay precision and accuracy were assessed by measuring the recovery of quality control samples (QCs) at concentrations of 50, 400, and 800 ng/ml. QCs were prepared separately from standards, aliquoted, and stored at −20°C. Standards and QCs were run on each microplate, and the recovery of QCs was calculated using the equation % recovery = 100 × calculated concentration using the standard curve/actual concentration of the sample. To assure the precision and accuracy of results, measured 8E3 concentrations were accepted only if the recovery of QCs fell in the range of 80%–120%, and if the CV values were less than 30% for all standards and QCs. A representative standard curve and the assay validation results are shown in Supplemental Fig. 1 and Supplemental Table 1, respectively.
A typical indirect ELISA scheme was applied in the assay to detect binding of 8E3 to K2 capsular polysaccharide. Briefly, 3 µg/ml K2 capsular polysaccharides prepared in 20 mM Na2HPO4 were used as the coating antigen. Nunc Maxisorp 96-well plates (Thermo Fisher Scientific) were coated with 100 µl/well antigen and incubated at room temperature for 2 hours. Plates were washed three times with 0.05% Tween-20 in 20 mM Na2HPO4, followed by three washes with distilled water. Samples were diluted 100× in PBS to minimize matrix interference. Diluted samples, standards, and QCs were added in triplicate (100 µl/well) and incubated for 2 hours. The plate was rewashed as described and 1000× diluted goat anti-mouse IgG-alkaline phosphatase conjugate (100 µl/well) (Thermo Fisher Scientific) was added followed by incubation for 1 hour. Finally, 200 µl of 4 mg/ml p-nitrophenyl phosphate in diethanolamine substrate buffer (Thermo Fisher Scientific) was added to each well, and change of absorbance over time (dA/dt) at 405 nm was measured using a microplate reader (SpectraMax 340PC; Molecular Devices, San Jose, CA). A similar ELISA method was developed to measure the mIgG3 isotype control (mIgG3; Novus Biologicals) in the antibody stability assay. The mIgG3 control binds keyhole limpet hemocyanin.
Effect of Iodine Labeling on the Binding of 8E3 to the K2 Capsule
8E3 was labeled with iodine-127 (127I) using chloramine-T method as described by Garg and Balthasar (2007). A competitive ELISA was used to measure the equilibrium dissociation constant (Kd) of 8E3. A Nunc Maxisorp 96-well plate was coated with 1 µg/ml isolated K2 capsular polysaccharide (100 µl/well) in 20 mM Na2HPO4 at room temperature for 2 hours. The plate was washed with 0.05% Tween-20 in 20 mM Na2HPO4. One hundred microliters of 8E3 or 127I-8E3 were added to each well at a concentration range of 0.01–10,000 nM, with 500× diluted 8E3-alkaline phosphatase conjugates in each sample. The plate was incubated for 2 hours and rewashed as described. Substrate p-nitrophenyl phosphate was then added at 4 mg/ml and 200 µl/well, and dA/dt at 405 nm was measured using a microplate reader. Data were fitted in the equation:

(Bmax = binding capacity; C = antibody concentration; R = dA/dt), and Kd values were estimated using Phoenix WinNonlin 7.0.
Endotoxin Quantification and Removal
Endotoxin in purified mAb 8E3 was quantified and removed using ToxinSensor chromogenic LAL endotoxin assay kit (GenScript, Piscataway, NJ) and Pierce high-capacity endotoxin removal spin columns (Thermo Fisher Scientific), respectively. The removal of endotoxin procedure was repeated to ensure that the endotoxin concentration in antibody preparation was ≤1 endotoxin units (EU)/ml.
Animal Studies
All animal studies were approved and conducted according to the Institutional Animal Care and Use Committee of the University at Buffalo and the Veterans Administration, in compliance with the recommendations in the Guide for the Care and Use of Laboratory Animals and the Radiation Safety Committee of the University at Buffalo. Female BALB/c mice were used for the development of monoclonal antibodies, and male BALB/c mice were used in all animal survival and antibody pharmacokinetic studies.
Mouse Pulmonary Challenge Model.
A murine pneumonia infection model as described previously by Russo et al. (2015) was used to assess the in vivo efficacy of 8E3. Briefly, male BALB/c mice, 7–8 weeks old, were anesthetized with isoflurane and hung on a silk suture by their maxillary incisors. Next, the tongue underwent gentle traction to prevent swallowing and ∼1 × 109 CFU of AB899 in 50 µl PBS was pipetted into the oropharynx to enable inoculation into lungs by reflexive aspiration. A dose of 50 or 200 mg/kg 8E3 (with or without endotoxin removal) or 200 µl of PBS (negative control) was injected intraperitoneally immediately after bacterial inoculation. To measure blood and organ bacterial burdens at 24 hours after the infection, mice were sedated with isoflurane and blood was collected via cardiac puncture. The vasculature then was perfused with 10 ml of PBS to remove residual blood in organs. Lungs and spleens were harvested and homogenized using a Bullet Blender Storm (Next Advance, Inc., Troy, NY). Bacterial burdens in blood, lung, and spleen were quantified by 10-fold serial dilution. 8E3 concentrations in isolated plasma samples were measured via ELISA (described above). Five mice were euthanized at each time point to collect blood and tissue samples.
Plasma Pharmacokinetics of 8E3 in Noninfected Mice.
To evaluate the plasma pharmacokinetics in noninfected mice, 8E3 was labeled with iodine-125 (125I) using the chloramine-T method as described by Garg and Balthasar (2007). Male BALB/c mice, 7–8 weeks old (n = 5/dose group), were anesthetized with isoflurane and administered 8E3 at doses of 1, 10, and 25 mg/kg that contained a trace amount of 125I-8E3 (400 µCi/kg 125I activity) via penile vein injection (five mice per dose group). Blood samples (40 µl) were collected from the saphenous vein at 1, 3, 8 hours, and 1, 3, 7, 10, and 14 days postdosing. An aliquot of whole blood was retained and plasma was separated by centrifugation at 2000g for 10 minutes. Proteins in plasma samples were precipitated by the addition of 200 µl of 1% bovine serum albumin in PBS and 500 µl of 10% trichloroacetic acid (TCA) in PBS to each sample. Plasma-TCA mixtures were allowed to incubate on ice for 15 minutes. TCA-precipitated proteins were then pelleted via centrifugation at 14,000g for 5 minutes, and the precipitates were washed three times with 700 µl of PBS. Radioactivity in protein pellets was assessed using a gamma counter (LKB Wallac Model 1272, Mt Waverley, Victoria, Australia), and radioactive counts in samples were corrected for background and decay. A group of five mice was intravenously injected with unlabeled 8E3 at 25 mg/kg, and the plasma 8E3 concentrations were measured using the developed ELISA.
Plasma Pharmacokinetics and Tissue Distribution of 8E3 in Noninfected and Infected Mice.
The murine pneumonia model was used to investigate 8E3 tissue distribution in AB899-infected mice. In brief, male BALB/c mice were challenged with ∼1 × 109 CFU of AB899 in 50 µl of PBS. Infected animals were then immediately injected intraperitoneally with 8E3 at doses of 50, 200, and 500 mg/kg containing 400 µCi/kg of 125I-8E3 (five mice per dose group), and another group of five noninfected mice was dosed with 125I-8E3 at 200 mg/kg. Blood samples were collected at 1, 3, 8, and 24 hours postdosing. Tissue samples from the lung, heart, liver, spleen, kidney, small intestine, skin, and muscle were harvested at 24 hours. Radioactive counts in blood and tissue samples were measured using a gamma counter and values were normalized by sample weights. 8E3 concentrations in all tissue samples were corrected for residual blood in the tissue based on the method described by Garg and Balthasar (2007). Plasma and blood 8E3 concentration data were analyzed using noncompartmental analysis in Phoenix WinNonlin 7.0 (Pharsight Corporation, St. Louis, MO). The following pharmacokinetic parameters were estimated from noncompartmental analysis: Cmax, area under the concentration-time curve (AUC), and t1/2. All other data were analyzed using GraphPad Prism 7.00 (GraphPad Software, Inc., San Jose, CA).
Another group of five infected mice were injected intravenously with 50 mg/kg 125I-8E3 to assess the antibody bioavailability of intraperitoneal injection. Blood samples were collected at 1, 3, 8, and 24 hours postdosing. Radioactive counts in blood samples were measured to calculate 8E3 concentrations as described above. The AUC of blood 8E3 was calculated to estimate the bioavailability (%F) based on the equation %F = 100 × AUCIP, 50 mg/kg/AUCIV, 50 mg/kg, where IP is intraperitoneal and IV is intravenous.
Stability of 8E3 in A. baumannii Culture
To investigate the stability of 8E3 in bacterial culture, the A. baumannii strains AB307-0294, AB899, HUMC6, HUMC1, AB985, and AB955 were used. Bacteria were grown in LB medium to mid-logarithmic phase, achieving an approximate titer of 1 × 107 CFU/ml, then 8E3 was added to either the bacterial culture or to the corresponding bacterial supernatants, followed by incubation at 37°C. Samples (20 µl) were collected at 1, 2, 3, and 6 hours to assess the 8E3 concentration using the developed ELISA. 8E3, incubated in 1 ml of sterile LB medium, served as a positive control, and anti–keyhole limpet hemocyanin mIgG3 was used as an isotype control. Where indicated, mAbs were incubated with sterile filtered (through 0.2 µm filters) bacterial supernatants, and samples were analyzed by ELISA and Western blot to assess 8E3 concentrations and antibody degradation, respectively. The sterility of bacterial supernatants was confirmed by plating on LB agar plates.
Bacterial Cell Adherence/Invasion Assay
Adherence and/or invasion of A. baumannii to human lung epithelial cells NCI-H292 were determined using the method described by Weiser et al. (2003) with modifications. Clinical isolates of A. baumannii (5 × 106 CFU) were incubated with 8E3 or F(ab′)2 of 8E3 at concentrations ranging from 13.3 to 333 nM for 60 minutes at room temperature. Opsonized bacteria were then added to a 24-well plate containing 5 × 105 NCI-H292 cells/well to give an MOI of 10:1. Plates were centrifuged at 1200g for 5 minutes and incubated at 37°C for 60 minutes. NCI-H292 cells were then washed five times with PBS and detached from the plates with 250 µl of 0.25% trypsin-EDTA (Thermo Fisher Scientific) and lysed by 0.5 mg/ml saponin to release intracellular bacteria. Serial 10-fold dilutions of samples were quantified by plating on LB agar plates. Experimental results are expressed as relative adherence/invasion compared with the CFU of mIgG3 isotype control.
Cytotoxicity Assay
Cell viability of NCI-H292 in bacterial adherence/invasion studies was measured using lactate dehydrogenase (LDH) cytotoxicity assay as described by the manufacturer (Thermo Fisher Scientific). Cell supernatants from the bacterial adherence assay were harvested and incubated with substrate mix (containing tetrazolium salt) at 37°C for 30 minutes. Released LDHs from dead NCI-H292 cells were then quantified by formazan formation (absorbance at 490 nm). Percentage cytotoxicity = 100 × (sample value − NCI-H292 spontaneous release − bacterial spontaneous release)/(NCI-H292 maximum release − NCI-H292 spontaneous release).
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 7.00 (GraphPad Software, Inc.). The Student’s t test was used to analyze bacterial burdens in mouse tissue samples, and blood/plasma and tissue concentrations of 8E3. The survival of mice was analyzed using log-rank (Mantel-Cox) test. The results of opsonophagocytosis and bacterial adherence assays were analyzed using one-way analysis of variance (ANOVA) followed by Dunnett’s multiple-comparison test. Two-way ANOVA followed by Dunnett’s multiple-comparison tests were performed to analyze in vitro 8E3 stability data.
Results
Development of mAbs Targeting the K2 Capsular Polysaccharide of A. baumannii.
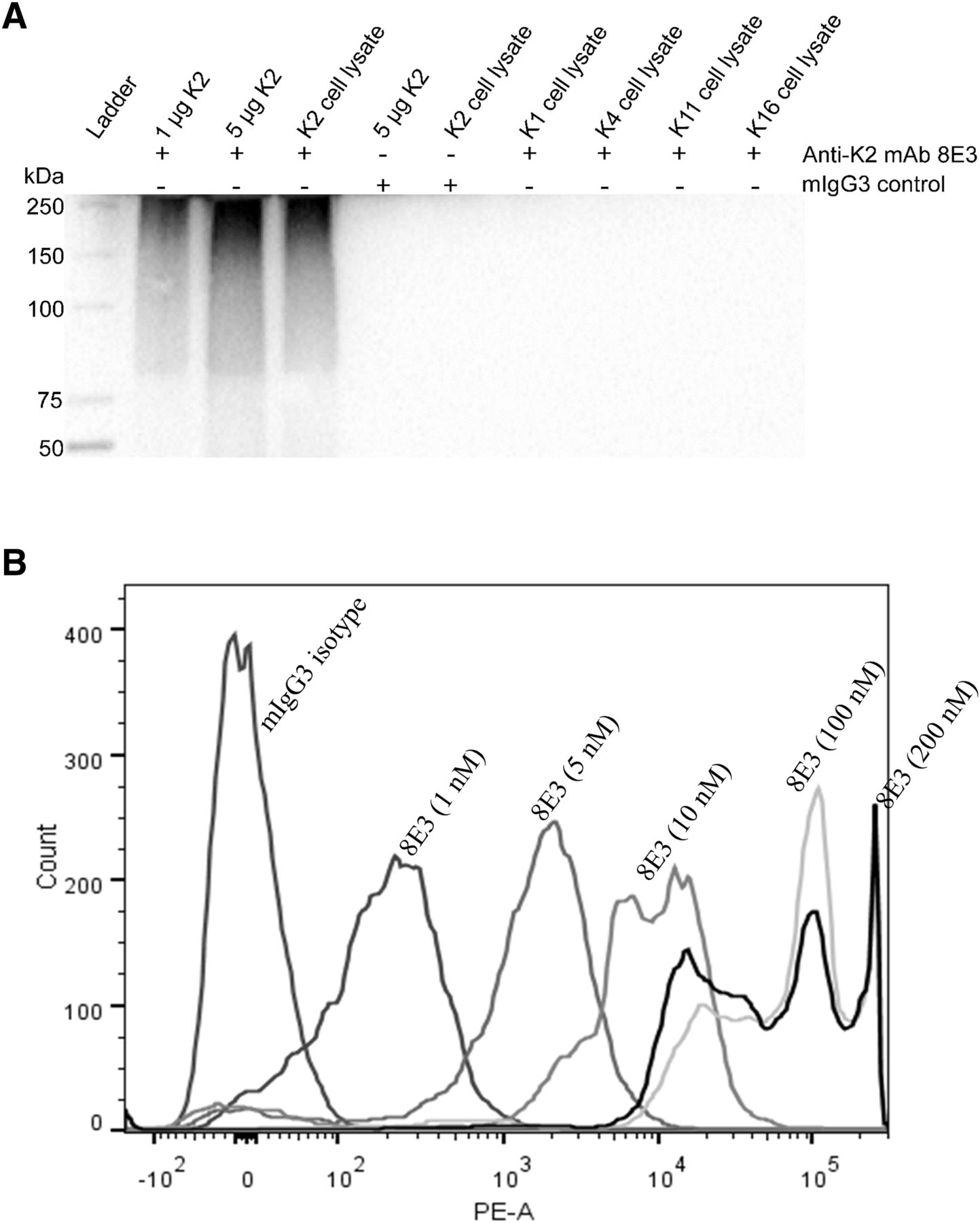
An mIgG3 (κ) mAb, 8E3, with specificity for the K2 capsular polysaccharide of A. baumannii, was developed via immunizing mice with isolated K2 capsular polysaccharides. The binding specificity of 8E3 to K2 capsules was assessed via Western blot analysis of isolated K2 capsules and whole-cell lysates of A. baumannii capsule types K1, K2, K4, K11, and K16 (Wang-Lin et al., 2017). Positive binding to isolated K2 capsule and K2 whole-cell lysates but not in cell lysates of other capsule types confirmed the specificity to the K2 capsule (Fig. 1A). 8E3 exhibited concentration-dependent binding to A. baumannii when evaluated with the K2 capsule type pathogen AB899. Geometric mean fluorescence intensity, measured via flow cytometry, increased as 8E3 concentration increased from 1 to 100 nM. Saturation of binding was observed at concentrations ≥100 nM, in which mean fluorescence intensity shifted ∼100,000-fold compared with the mIgG3 isotype control (Fig. 1B).

Binding of anti-K2 capsule mAb 8E3 to isolated K2 capsular polysaccharides and AB899. (A) Western blot analysis of isolated K2 capsules and whole-cell lysates of A. baumannii capsule types K1, K2, K4, K11, and K16. Bacterial cell lysates were prepared by sonication, as described by Cuenca et al. (2003). K2 capsules and bacterial cell lysates were electrophoresed in a 4%–15% polyacrylamide gel. Binding of 8E3 to K2 capsules was assessed on a polyvinylidene difluoride membrane, detected by anti-mouse IgG horseradish peroxidase conjugates, and visualized using enhanced chemiluminescence substrates. (B) 1 × 106 CFU of AB899 were incubated with 8E3 at a concentration range of 1–200 nM for 60 minutes. Binding of antibodies to bacteria was detected by goat anti-mouse IgG-R-phycoerythrin conjugates and measured using flow cytometry. A commercial mIgG3 (Novus Biologicals) was used as an isotype control for the detection of nonspecific binding. PE-A, phycoerythrin area.
Anti-K2 Capsule mAb 8E3 Augmented Phagocytic Killing of A. baumannii In Vitro.
An anti-K1 capsule IgM, 13D6, developed previously in the laboratory of T.A.R., has been demonstrated to enhance neutrophil-mediated bactericidal activity when tested against K1 capsule type A. baumannii strains in vitro (Russo et al., 2013; Wang-Lin et al., 2017). Thus, we hypothesized that opsonization with anti-K2 capsule mAb 8E3 would increase macrophage-mediated killing of AB899. Opsonophagocytosis of AB899 was assessed using both J774A.1 and RAW 264.7 macrophage cells at a wide range of MOIs, from 1:6 to 312:1, bacteria to macrophages. This range of MOI included 105–108 CFU/ml AB899, which represents tissue bacterial burdens at different stages of A. baumannii infection. Incubation of 8E3-opsonized AB899 with J774A.1 for 60 minutes at 37°C resulted in a maximum of ∼75% reduction in bacterial CFU at 6.67 nM (1 µg/ml) 8E3 when ∼105 CFU/ml AB899 (MOI, 1:3) were added in the assay (Fig. 2A). Higher antibody concentrations were required to reach a similar magnitude of bactericidal activity when greater bacterial concentrations were used. Preopsonization with 33.3 nM (5 µg/ml) and 133 nM (20 µg/ml) 8E3 led to ∼90% and ∼65% killing of bacteria when 106 CFU/ml (MOI, 19:1) and 107 CFU/ml (MOI, 2:1) AB899 were used, respectively (Fig. 2A). A similar antibody concentration–dependent phagocytic killing of AB899 was observed when evaluated using RAW 264.7 cells with MOIs of 1:6 (Fig. 2B) and 312:1 (Fig. 2C). These results suggest that capsular polysaccharide could be a potential therapeutic target for A. baumannii infection. These data also imply the importance of early treatment since a larger dose of antibody may be needed for optimal efficacy with bacterial growth and progression of infection.

Opsonization with mAb 8E3 significantly enhanced murine macrophage-mediated phagocytic killing of AB899. Bacteria were incubated with 8E3 at concentrations range from 6.67 to 6667 nM (i.e., 1–1000 µg/ml) for 60 minutes. The opsonized AB899 were then exposed to 5 × 105 murine macrophage cells J774A.1 or RAW 264.7 in a 24-well plate to give an MOI (bacteria to macrophages) of 1:6 to 312:1. Antibody concentration and MOI-dependent phagocytic killing of AB899 was observed with J774A.1 cells at MOI of 1:3, 2:1, and 19:1 (A), and with RAW 264.7 cells at MOI of 1:6 (B) and MOI of 312:1 (C). Results were expressed as the percentage of survival compared with the bacterial CFU of a mIgG3 isotype control. n = 3–4. ***P < 0.001; ****P < 0.0001 (one-way ANOVA followed by Dunnett’s multiple comparisons). The data are presented as the mean ± S.D.
Anti-K2 Capsule mAb 8E3 Enhanced A. baumannii Infection in a Murine Pneumonia Infection Model.
Opsonization with anti-K2 capsule mAb 8E3 showed potent macrophage-mediated bactericidal activity against AB899 at a wide MOI range of 1:6–312:1, suggesting a potential therapeutic utility of 8E3. To explore the potential efficacy of 8E3 in animals, mice underwent pulmonary challenge with AB899 at an inoculum of ∼1 × 109 CFU (Russo et al., 2015). Fifty milligrams per kilogram 8E3 was injected immediately after bacterial infection, which was estimated to provide plasma concentrations at least 10-fold higher than the Kd value of 8E3. Unexpectedly, 8E3 treatment did not improve the animal survival compared with the PBS control group (Fig. 3A). Further and surprisingly, free 8E3 (i.e., target-unbound 8E3) concentrations were undetectable in all mouse plasma samples collected at 24 hours as determined by ELISA (see Materials and Methods for assay development) (Fig. 3B). In addition, a significantly higher blood bacterial load (3.05-fold in mean) was observed in mice treated with 8E3 compared with mice treated with PBS (Fig. 3C). These results prompted another trial in which mice were treated with a 4-fold greater dose of 8E3 (200 mg/kg). However, again, significant increases in the bacterial load of AB899 and in mortality were observed. All mice treated with 8E3 succumbed to infection by 48 hours, whereas PBS-treated mice had 100% survival at 48 hours and 30% survival at 72 hours (Fig. 3D). Plasma unbound 8E3 concentrations were again below the LLOD in all mice despite treatment with this higher 8E3 dose (Fig. 3E). Additionally, at 24 hours postinfection, mean bacterial burdens in lung, blood, and spleen increased by 6.64-fold, 9.58-fold, and 253-fold, respectively, compared with PBS-treated mice (Fig. 3F). First, these results suggest that 8E3 either underwent rapid elimination (e.g., potentially mediated by bacterial enzymatic degradation or via target-mediated elimination) or 8E3 was bound to a targets with an extremely large binding capacity. Second, the increased bacterial load and mortality observed in 8E3-treated mice suggests that 8E3 treatment leads to ADE of A. baumannii infection.

Passive immunization with mAb 8E3 was associated with enhanced infection of AB899 in a mouse pneumonia model. BALB/c mice underwent pulmonary challenge with AB899 and were immediately treated intraperitoneally with 50 mg/kg (A–C) or 200 mg/kg (D–F) 8E3 (in 200 µl of PBS) or 200 µl of sterile PBS. (A–C) Animals were infected with 1.23 × 109 CFU of AB899 (n = 10/group). (D–F) Mice were infected with 1.12 × 109 CFU of AB899 (n = 15/group). (A and D) Animal survival was analyzed using Log-rank (Mantel-Cox) test. (B and E) Plasma-free 8E3 concentrations at 24 hours after the injection were undetectable in both 8E3- and PBS-treated mice using the developed ELISA. Recoveries of (QCs were within the range of 91%–105% with CV less than 25%. LLOQ was 50 ng/ml in 100× diluted mouse plasma. The data are presented as the mean ± S.D. (C and F) Bacterial burdens in blood, lung, and spleen at 24 hours after infection were analyzed using two-tailed unpaired t test. The data are presented as the mean ± S.D. *P < 0.05; **P < 0.01; ****P < 0.0001.
Plasma Pharmacokinetics of 8E3 in Noninfected Mice Excluded Off-Target Binding.
To assess the potential for nonspecific or off-target antibody binding, which may contribute to the rapid reduction of unbound 8E3 concentrations in mouse plasma, the plasma pharmacokinetics of 8E3 were studied in noninfected mice at intravenous doses of 1, 10, and 25 mg/kg with a trace amount of 125I-8E3. 8E3 concentrations were quantified via measuring the radioactivity of 125I-8E3. First, to confirm that iodine labeling did not affect the 8E3 binding to its target antigen, the Kd of 127I (nonradioactive) labeled 8E3 was measured and compared with the Kd of unlabeled 8E3 using a competitive ELISA. Binding profiles of the 127I-8E3 and 8E3 nearly overlapped, and the estimated Kd values of 127I-8E3 and 8E3 were 301 and 430 nM, respectively. Therefore, iodine labeling did not adversely affect the binding of 8E3 to the K2 capsular polysaccharide. Second, the potential effect of 125I labeling on the pharmacokinetics of 8E3 was assessed by comparing the plasma 8E3 concentrations measured using ELISA to those assessed via 125I. Similar 8E3 concentrations were found (Fig. 4A) indicating that 125I labeling did not impact the pharmacokinetics of 8E3 in mice. Typical biexponential antibody concentration-time profiles were observed for plasma 8E3 at all studied doses (Fig. 4B). The t1/2 of 8E3 was estimated to be 6.74–8.03 days, which is consistent with the typical mIgG3 t1/2 of 6–8 days (Vieira and Rajewsky, 1988). Plasma 8E3 concentration-time profiles overlapped when normalized to injected doses (Fig. 4C), indicating linear pharmacokinetics in noninfected mice at the dose range of 1–25 mg/kg. Therefore, 8E3 exhibited typical linear pharmacokinetics of an mIgG3 in noninfected mice, and nonspecific/off-target binding that could impact the 8E3 elimination and t1/2 was deemed unlikely.

Plasma concentration (Cp)-time profiles of 8E3 after a single intravenous dose of 8E3 in noninfected mice. (A) Plasma 8E3 concentrations measured using ELISA and the 125I labeling method after a single intravenous dose of 25 mg/kg. Plasma 125I-8E3 concentration-time profiles presented for each 8E3 dose at 1, 10, and 25 mg/kg (B), and as dose-normalized concentration-time profiles (C). n = 5. The data are presented as the mean ± S.D.
Decreased Concentration of Free 8E3 after Exposure to AB899 Was Consistent with Binding to Capsular Polysaccharide, Not Enzymatic Degradation.
Several bacterial species have been found to secrete proteinases that degrade Igs, such as IgG-degrading enzyme of Streptococcus pyogenes (IdeS) and Streptococcal erythrogenic toxin B (SpeB), which cleave the γ-chain of human IgG in the hinge region and hence assist bacterial evasion of antibody-dependent effector functions (Collin and Olsén, 2001; von Pawel-Rammingen et al., 2002). A. baumannii also could express IgG-specific proteinases that may lead to the degradation of 8E3, although such proteinases have not been reported. To investigate potential enzymatic degradation of 8E3 by A. baumannii, 100 µg/ml 8E3 was incubated in bacterial cultures containing ∼1 × 107 CFU of various A. baumannii strains or in the corresponding bacterial supernatants, including strain AB307-0294 (capsule type, K1), strains AB899 and HUMC6 (capsule type, K2), strain HUMC1 (capsule type, K4), strain AB985 (capsule type, K11), and strain AB955 (capsule type, K16). Unbound 8E3 concentrations dropped below the ELISA LLOD after incubation for 1 hour with A. baumannii K2 strains AB899 and HUMC6, whereas 8E3 concentrations did not change substantially over time compared with LB medium control when evaluated in cultures of non-K2 capsule–type strains (Supplemental Fig. 2A). These data show that the A. baumannii–mediated decrease in free 8E3 concentration is K2 strain specific (potentially relating to target binding or K2 strain–unique proteinases). Concentrations of an mIgG3 (50 µg/ml) isotype control did not change significantly in all tested A. baumannii cultures over time (Supplemental Fig. 2B), consistent with the absence of IgG3 proteinases.
This finding was further supported when 8E3 was incubated with sterile filtered supernatants of mid-logarithmic A. baumannii cultures. Decreases in free 8E3 concentrations (∼60% reduction) were found to be K2 capsule type specific (consistent with binding to shed K2 capsules) and time-independent (consistent with absence of enzymatic degradation) (Supplemental Fig. 2C). Additionally, Western blot analysis of the 8E3 supernatant samples at 3 hours did not demonstrate 8E3 degradation. Antibody bands detected after 8E3 was exposed to bacterial supernatants (lanes 5–10) were the same as those after 8E3 was exposed to sterile LB medium (lane 2) (Supplemental Fig. 2D) and unadulterated 8E3 (Supplemental Fig. 2E). Bands at 150 kDa were intact 8E3 molecules with different levels of glycosylation, and light bands at 100 and 75 kDa were likely antibody fragments generated during purification. Therefore, these data support the hypothesis that decreases in free 8E3 after exposure to live and culture supernatants of A. baumannii K2 strains were due to binding of 8E3 to cell-associated and/or shed capsular polysaccharides, but not to enzymatic degradation.
The K2 Capsular Polysaccharide Has a High Capacity for Binding 8E3.
The binding capacity of the K2 capsule was estimated in vitro, in which 10–100 µg/ml 8E3 was incubated with 1 × 107 CFU/ml AB899 culture (i.e., bacteria and supernatant-containing shed capsules) or AB899 bacteria (i.e., bacteria washed with PBS) at room temperature for 30 minutes. Unbound 8E3 concentrations were measured by ELISA. The binding capacities of 1 × 107 CFU/ml AB899 culture and AB899 bacteria were estimated to be 77.3 and 24.4 µg/ml, respectively. Thus, the shed capsules in the corresponding AB899 culture were able to bind 52.9 µg/ml 8E3 (i.e., 77.3–24.4 µg/ml). Namely, 1 CFU of AB899 and its shed capsules can bind ∼1 × 107 and 2.12 × 107 8E3 molecules, respectively (binding capacity = remaining 8E3 amount/IgG molecular mass × Avogadro constant/1 × 107 CFU). The surprisingly large binding capacity of shed and bacterially associated capsule likely resulted in the observed rapid decrease of unbound 8E3 concentrations in mouse plasma.
Effect of A. baumannii Infection on Blood Pharmacokinetics and Tissue Distribution of 8E3.
125I-labeled 8E3 was used to quantify the total antibody concentrations in mouse blood and tissues, in view of undetectable free 8E3 concentrations as measured by ELISA in infected mice. This methodology measures bound and unbound antibody, without the requirement of antigen binding for detection, whereas mAb quantification via ELISA measures only unbound antibody. Mice were challenged with 1 × 109 CFU of AB899 and were immediately treated intraperitoneally with 50, 200, and 500 mg/kg 8E3. Blood 8E3 concentration-time profiles for each antibody dose are shown in Fig. 5A. Cmax 8E3 values were observed at 3 hours postdosing and demonstrated a dose-proportional increase from 1.16 × 102 to 5.10 × 102 to 1.29 × 103 µg/ml (Table 1). Dose-normalized blood 8E3 concentration-time profiles nearly overlapped (Fig. 5B), suggesting linear blood pharmacokinetics in the dose range of 50–500 mg/kg.

Blood concentration (Cb)-time profiles of 8E3 after a single intraperitoneal dose of 8E3 in mice that were intratracheally infected with 1 × 109 CFU of AB899. Blood 8E3 concentrations were quantified by measuring the radioactivity of 125I-8E3 in blood samples. (A) Data are shown for each 8E3 dose at 50, 200, and 500 mg/kg. (B) Dose-normalized 8E3 Cb-time profiles. n = 5. The data are presented as the mean ± S.D.
Noncompartmental analysis of blood 8E3 concentrations after a single dose of 8E3 in noninfected and AB899-infected mice
Data are presented as the mean ± S.D. (n = 5).
Infection with AB899 did not impact the blood 8E3 pharmacokinetics when mice were treated intraperitoneally with 200 mg/kg 8E3, as demonstrated by overlapping blood 8E3 concentration-time curves (Fig. 6A) and similar values of Cmax (5.10 × 102 vs. 5.60 × 102 µg/ml) and AUC for 0–24 hours (AUC0–24 h) for blood 8E3 (8.70 × 103 vs. 8.28 × 103 h/µg per milliliter) between infected and noninfected mice (Table 1).

Blood and tissue distributions of 8E3 after a single intraperitoneal dose of 200 mg/kg 8E3 in mice infected with 1 × 109 CFU of AB899 or 50 µl sterile PBS. 8E3 concentrations were measured using 125I-labeling method. (A) Blood concentration (Cb)-time profiles of 8E3 in infected (filled symbols) and noninfected (open symbols) mice. (B) Cb and plasma concentration (Cp) of 8E3 at 24 hours in infected and noninfected mice. Mean values of the Cb/Cp ratio were shown above the bars. 8E3 concentrations (C) and the amount of 8E3 expressed as a percentage of the injected dose (D), in mouse blood and tissues that were harvested at 24 hours postdosing. Filled bars are infected mice, and open bars represent noninfected mice. n = 5. *P < 0.05; ***P < 0.001; ****P < 0.0001 (two-tailed unpaired t test). The data are presented as the mean ± S.D. S intestine, small intestine.
Blood instead of plasma 8E3 concentrations were measured in this study because antibody concentrations in plasma may be underestimated because of the removal of soluble complexes of shed capsule and antibody, and/or complexes of 8E3-A. baumannii from centrifugation during the process of plasma isolation (as discussed below in the formation of high-order antibody-bacteria complexes). This concern was validated by the determination of the mean ratio of blood to plasma 8E3 concentrations at 24 hours postdosing in noninfected and infected mice; the ratio was 0.567 in noninfected mice, which is close to expectations (0.55) based on a typical hematocrit (i.e., plasma volume is typically 55% of blood volume) but was 1.97 in infected mice (Bonate and Howard, 2004), indicating that a substantial fraction of 8E3 is present in complexes (e.g., with soluble capsule and/or A. baumannii) (Fig. 6B). Therefore, pharmacokinetic calculations based on plasma antibody concentrations in bacterium-infected animals can lead to biased values (overestimation) of antibody clearance, as observed in this case.
Although 8E3 blood pharmacokinetics were not different between infected and noninfected mice, A. baumannii infection significantly influenced the distribution of 8E3 in tissues, especially in tissues associated with high quantities of bacteria (e.g., lung, liver, and spleen). Antibody concentrations at 24 hours postdosing in tested tissues were corrected for the residual blood as described previously, where volumes of residual blood are estimated through the use of chromium-51–labeled red blood cells (Garg and Balthasar, 2007). 8E3 distributions in tissues are shown in Fig. 6, C and D, presented as 8E3 concentrations and the percentage of the injected dose. Significant amounts of 8E3 accumulated in the lung, liver, spleen, kidneys, and small intestine in AB899-infected mice compared with the tissues in noninfected mice (Table 2). These results suggest that bacteria mediated the accumulation of anti-K2 capsule mAb 8E3 in infected tissues, likely through the combined effects of vascular disruption and binding to bacterial capsule in infected issues. However, the antibody accumulation in infected tissues did not substantially impact the blood 8E3 pharmacokinetics in this experiment (Fig. 6A). Further, the bioavailability of 8E3 after intraperitoneal dosing was estimated to be 73.3% based on the AUC0–24 h ratio of an intraperitoneal dose to an intravenous dose of 8E3 at 50 mg/kg (Table 1). Of note, this value of bioavailability is likely an underestimate, because AUC data were collected for only 24 hours, and considering the slow absorption of antibody from peritoneal cavity and long 8E3 t1/2 values of 6.74–8.03 days. Prolonged sampling in infected mice was not possible, as animals succumb to infection ∼48 hours after bacterial inoculation.
Tissue distribution of 8E3 at 24 h after a single intraperitoneal dose of 200 mg/kg 8E3 in noninfected and AB899-infected mice
Data are presented as the mean ± S.D. (n = 5).
Endotoxin Contamination Was Not Responsible for ADE of A. baumannii Infection in Mice.
Septic shock caused by lipopolysaccharide is a major cause of death among patients with bacterial infections (Parrillo, 1993). Khan et al. (2000) have shown intravenous injection of 1 × 106 and 1 × 107 U of endotoxins resulted in 0% and 100% mortality in BALB/c mice, respectively. The residual endotoxin concentration in purified mAb 8E3 was relatively low, but the concentration was determined to be 1.07 × 103 EU/ml in the concentrated preparation (i.e., 20 mg/ml antibody). Injection of 200 µl of this antibody preparation yields ∼200 EU/ml endotoxin in mouse plasma. Although this quantity of endotoxin is not sufficient to cause animal death, we considered the possibility that it may have increased the release of cytokines, leading to septic shock. To exclude the possible contribution of adverse effects from coadministered endotoxins in antibody preparations, the mouse survival study was repeated with endotoxin-free (defined as an endotoxin concentration of ≤1 EU/ml in this study) antibody and PBS preparations. Endotoxins were removed using a polylysine affinity column, and endotoxin concentrations were reduced to 0.83 and 0.25 EU/ml in 8E3 and PBS preparations, respectively. Similar results were found in the repeat investigation, confirming that 8E3 treatment contributed to the increased growth of A. baumannii and mouse mortality. All mice treated with endotoxin-free 8E3 died within 48 hours, whereas 80% of mice survived for ≥72 hours in PBS group (Fig. 7A). Antibody-treated mice were found to have significantly higher amounts of AB899 in lung samples, and with an average of 21.1-fold and 490-fold higher bacterial counts, respectively, in blood and spleen samples, compared with bacterial counts found in PBS-treated mice (Fig. 7B). Therefore, similar to previous results, treatment with endotoxin-depleted anti-K2 capsule mAb 8E3 did not confer protection against AB899 infection, but, conversely, enhanced the growth of A. baumannii in mice. Of note, the most dramatic increase of bacterial load in both studies (i.e., treatment with non–endotoxin-depleted 8E3 and endotoxin-depleted 8E3) was observed in splenic tissue. These results were surprising given the observed opsonophagocytic killing of A. baumannii in vitro and the fact that the spleen is replete with mononuclear phagocytes; thus, the expectation would be increased bactericidal activity and hence decreased splenic bacterial burden with 8E3 treatment compared with treatment with PBS.

Passive immunization with endotoxin-free 8E3 enhanced the infection of AB899 in the mouse pneumonia model. BALB/c mice were challenged with 6 × 108 CFU of AB899 and were immediately treated intraperitoneally with 200 mg/kg endotoxin-free 8E3 or 200 µl of sterile endotoxin-free PBS (n = 10/group). Animal survival was analyzed using the log-rank (Mantel-Cox) test (A), and bacterial burdens in blood, lung, and spleen at 24 hours after infection were analyzed using the two-tailed unpaired t test (B). *P < 0.05. The data are presented as the mean ± S.D.
Anti-K2 Capsule mAb 8E3 Enhanced A. baumannii Adherence/Invasion to Human Lung Epithelial Cells via IgG Engagement of Fc Gamma Receptors.
Although ADE of bacterial infection is not common and the corresponding mechanisms remain unclear, ADE of viral infection has been more commonly reported and is typically mediated through antibody engagement of Fc receptors or complement receptors, which results in enhanced viral invasion to host cells. To investigate whether A. baumannii employs a similar mechanism, AB899 adherence and/or invasion of NCI-H292 human lung epithelial cells was evaluated in the presence and absence of 8E3. This FcγR-expressing cell line was chosen in view of the significant increase in A. baumannii CFU in lung in 8E3-treated mice. Preopsonization with 8E3 significantly increased AB899 adherence/invasion to NCI-H292 cells in a concentration-dependent manner. NCI-H292 cell–associated bacteria (i.e., bacteria attached to the epithelial cell surface and bacteria inside the cells) increased with 8E3 concentration from 0 to 133 nM (i.e., 0–20 µg/ml), and the increase in cell-associated bacteria (compared with bacterial CFU of mIgG3 isotype control group) reached a maximum of ∼10-fold at an 8E3 concentration of 133 nM (Fig. 8A). In addition, this 8E3 concentration-dependent enhancement of bacterial adherence/invasion to NCI-H292 was also found when other K2 strains were assessed, including the carbapenem-resistant clinical isolates obtained from Harbor-UCLA Medical Center (strain HUMC6) (Luo et al., 2012), University Hospitals of Cleveland (strain AB8407), and Erie County Medical Center (strain “Kelly lung”), as shown in Fig. 8B. These results indicate that the anti-K2 capsule mAb 8E3-mediated enhancement of A. baumannii adherence/invasion to host cells is not bacterial strain specific but likely occurs with all K2 capsule–type strains.

The anti-K2 capsule mAb 8E3 enhanced A. baumannii adherence/invasion to human lung epithelial cells NCI-H292. A. baumannii K2 clinical isolates were opsonized with 8E3 at concentrations of 0, 13.3, 33.3, 66.7, 133, and 333 nM (i.e., 0, 2, 5, 10, 20, and 50 µg/ml) for 60 minutes. The opsonized bacteria (5 × 106 CFU) were allowed to react with 5 × 105 NCI-H292 cells in a 24-well plate for 60 minutes at 37°C. Cell-associated bacterial CFU (i.e., cell surface bound + intracellular bacteria) were measured, and the data were expressed as relative adherence/invasion compared with the mIgG3 isotype control. Four different K2 clinical isolates of A. baumannii AB899 (A), and HUMC-6, AB8407, and “Kelly lung” (B) showed a concentration-dependent increase of adherence/invasion to NCI-H292 (P values vary from 0.0001 to 0.0228 for 8E3 concentrations of 13.3–133 nM). n = 3–4. ***P < 0.001; ****P < 0.0001 (one-way ANOVA followed by Dunnett’s multiple comparisons). The data are presented as the mean ± S.D.
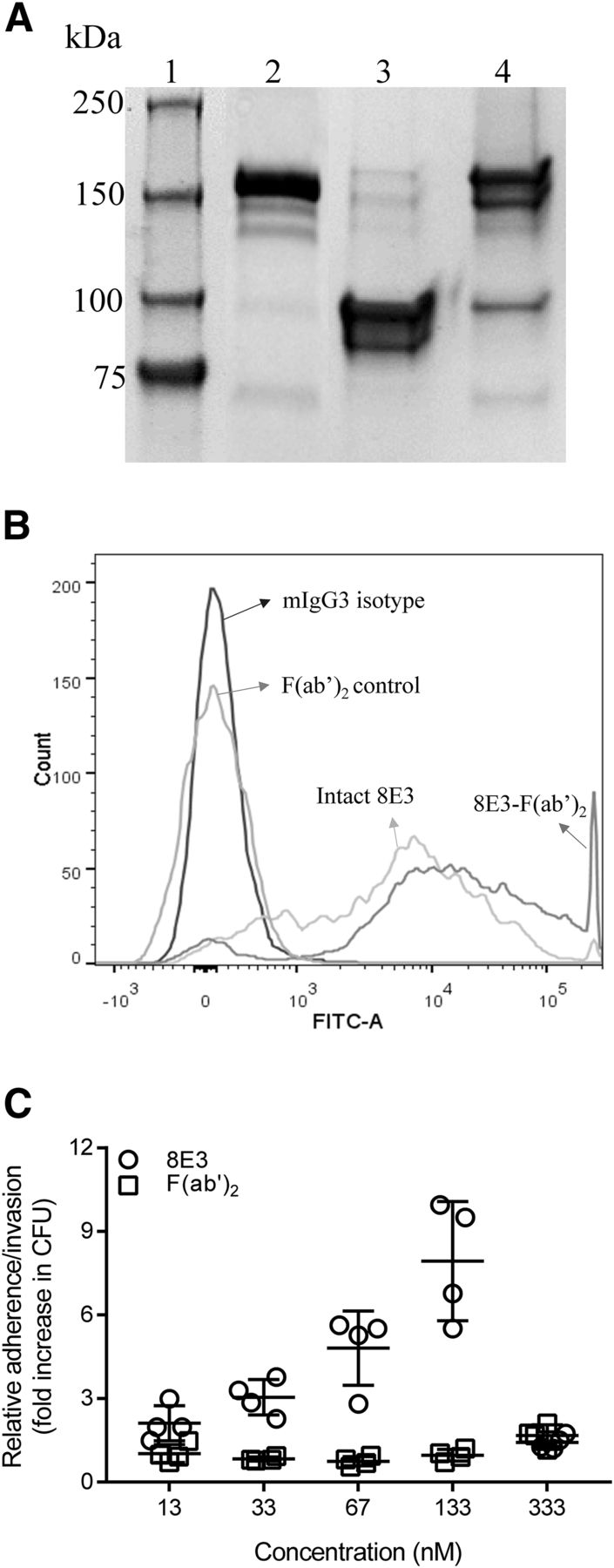
The potential mechanism for 8E3-mediated enhancement of A. baumannii adherence/invasion to host cells was also investigated. To examine for a possible role of the Fc domain, 8E3 was treated with pepsin to cleave Fc fragments and to generate F(ab′)2 fragments (∼100 kDa molecular weight in SDS-PAGE; Fig. 9A). Binding of F(ab′)2 to AB899 was not affected by pepsin digestion (Fig. 9B). Removal of the Fc domain from 8E3 via pepsin digestion abolished the antibody-mediated increase of AB899 adherence/invasion to human lung epithelial cells (NCI-H292) at all tested concentrations (Fig. 9C). Therefore, 8E3-enhanced adherence/invasion of A. baumannii K2 strains to human lung epithelial cells proceeds via IgG engagement of Fc gamma receptors.

Antibody 8E3–enhanced adherence/invasion of A. baumannii to human lung epithelial cells NCI-H292 through IgG engagement of FcγRs. (A) SDS-PAGE analysis of pepsin-digested 8E3; lanes 1–4 are ladder, 8E3, generated F(ab′)2, and undigested 8E3 eluate from protein A column, respectively. Undigested 8E3 in F(ab′)2 preparation is ∼3.5% based on the analysis using ImageJ (National Institutes of Health, Bethesda, MD). (B) Binding of F(ab′)2 to AB899 was not affected by pepsin digestion. Ten nanomolar antibody or F(ab′)2 was incubated with 1 × 106 CFU of AB899 for 60 minutes. Binding of antibody or F(ab′)2 to bacteria was detected by goat anti-mouse IgG (H+L)-FITC conjugates and was assessed using flow cytometry. (C) Opsonization of AB899 with 13–333 nM F(ab′)2 of 8E3 abolished the 8E3-mediated enhancement of AB899 adherence/invasion to NCI-H292 cells. Data were expressed as relative adherence/invasion compared with the bacterial CFU of mIgG3 isotype control. Statistically significant differences were not observed between mIgG3 control and the F(ab′)2 groups (one-way ANOVA followed by Dunnett’s multiple-comparison test). FITC-A, FITC area. The data are presented as the mean ± S.D.
The Decrease in Cell Associated A. baumannii at High 8E3 Concentrations (≥333 nM) Appears to be Due to the Formation of High-Order Antibody-Bacteria Complexes.
Interestingly, decreased relative adherence/invasion was observed at ≥333 nM (50 µg/ml) of 8E3 in NCI-H292 cells, for all tested A. baumannii strains (Fig. 8). This bell-shaped curve was also observed in S. pneumoniae when the anti-capsule IgA1-dependent enhancement of S. pneumoniae adherence was evaluated in human pharyngeal epithelial cells D562 (Weiser et al., 2003), but the reason for decreased relative adherence at high antibody concentrations has never been explored. Our in vitro studies found that the decreased relative adherence/invasion at high concentrations of 8E3 was not due to increased apoptosis of host cells, as demonstrated by negligible changes on NCI-H292 cell viability using LDH cytotoxicity assay (Supplemental Fig. 3), but it was likely due to the formation of high-order antibody-bacteria complexes (i.e., aggregates of two or more bacteria) (Supplemental Fig. 4, A and B). AB899 was incubated with 8E3 at concentrations ranging from 13.3 to 667 nM for 60 minutes at room temperature. The formation of antibody-bacteria complexes was assessed using flow cytometry, and the sizes of complexes were analyzed based on the forward scattering of particles. The percentage of formed higher-order antibody-bacteria complexes (i.e., particles with sizes two or more times the mean size of a bacterium) increased with 8E3 concentration and reached a maximum of ∼95% at ≥333 nM (Supplemental Fig. 4A, Q2 and Q3). The complexes were also visualized microscopically (Supplemental Fig. 4B), where the size and complexity of the antibody-bacteria complexes increased with 8E3 concentration. Therefore, the apparent decrease in relative adherence/invasion of A. baumannii at high 8E3 concentrations (≥333 nM) may be due to the formation of high-order antibody-bacteria complexes, as both a single bacterium and clusters of bacteria would appear to form a single colony on LB agar plates. Therefore, in some circumstances, studies including a narrow range of antibody concentrations or using the CFU quantification method may underestimate the potential antibody-mediated increase in bacterial adherence. Of note, the formation of higher-ordered complexes may be expected to be dependent on the relative concentrations of antibody and bacterial antigens. Concentrations of 1 × 106 and 5 × 106 CFU bacteria/ml were used in the in vitro antibody-bacterial complex formation (Supplemental Fig. 4) and in the bacterial adherence/invasion experiments (Fig. 8); however, bacterial concentrations in vivo ranged above 108–1010 CFU/ml in blood and lung samples obtained from infected mice. The potential formation of higher-order complexes in vivo, and the significance of this phenomenon, requires further investigation.
Discussion
The aims of the present study were to develop an mAb targeting the K2 capsular polysaccharide of A. baumannii and to assess its therapeutic potential in animals. An mIgG3 directed against the K2 capsular polysaccharide of A. baumannii was generated and exhibited potent bactericidal activity when evaluated in an opsonophagocytosis assay in vitro. However, not only did 8E3 treatment not provide protection against a clinical strain AB899 infection in a mouse pneumonia model, but 8E3 treatment substantially increased mouse mortality and bacterial burden in blood, lung, and spleen compared with the PBS control group. Based on our investigations, we propose that the lack of in vivo 8E3 efficacy and the apparent ADE of infection likely relate to the interaction of 8E3 with shed K2 capsule.
Initial investigations of 8E3 pharmacokinetics, as assessed with an antigen-specific indirect ELISA, an assay for unbound antibody, demonstrated an apparent rapid 8E3 elimination in infected mice, because 8E3 concentrations were below the LLOD (30 ng/ml) 24 hours after dosing of 50 and 200 mg/kg. However, subsequent investigations, using 125I-labeled 8E3, which measures bound and unbound antibody, demonstrated that 8E3 exhibited linear plasma pharmacokinetics at the dose range of 1–25 mg/kg in noninfected mice, with a typical mIgG3 t1/2 of 6.74–8.03 days. Although AB899 infection led to increased accumulation of 125I-8E3 in infected tissues such as the lung, liver, and spleen, blood 125I-8E3 pharmacokinetics were not significantly influenced by AB899 infection, and linear blood 125I-8E3 pharmacokinetics were observed at the dose range of 50–500 mg/kg in infected mice. Interestingly, substantially lower blood bacterial load was observed in mice treated with 200 mg/kg 8E3 compared with 50 mg/kg 8E3 (Fig. 3, C and F), suggesting that 8E3 may impact the disposition of AB899 either via the formation of higher-order antibody-bacteria complexes, or through increasing the bacterial adherence to and/or invasion of endothelial cells, or the accumulation of bacteria in lungs.
The pharmacokinetic results may be explained by the finding that shed K2 capsule has a large capacity to bind 8E3. Shed capsule from 1 × 107 CFU of AB899 in 1 ml of LB medium was estimated to bind 52.9 µg of 8E3; thus, 8E3 concentrations above 50 mg/ml may be required to neutralize shed capsule components in infected mouse lung if a 1 × 1010 CFU/ml bacterial burden is assumed. As such, even after the administration of a large dose of mAb (e.g., 200 mg/kg 8E3), plasma and tissue concentrations are well below the estimated binding capacity of shed capsule components. The observed 8E3 concentration in infected lung at 24 hours postdosing was 1379 ± 150 µg/ml (Table 2), which is 38.4-fold lower than the estimated antibody concentration required to neutralize shed K2 capsules (i.e., 52.9 mg/ml). Therefore, it is likely that shed capsules from A. baumannii act as “decoys” that neutralize the anti-K2 capsule mAb 8E3 and substantially reduce the amount of 8E3 reaching the bacterial surface, as needed for antibody-dependent bactericidal activity. In vitro opsonophagocytosis results also support these data, in which 1 mg/ml concentrations of 8E3 were needed to show substantial bacterial killing when evaluated against ∼1 × 108 CFU/ml AB899 (Fig. 2C). Of note, similar decoy effects of shed capsule have been observed in prior reports with other bacteria (e.g., Staphylococcus epidermidis, Klebsiella pneumoniae, S. pneumoniae, and Pseudomonas aeruginosa), although quantitative measurements were not reported (Cerca et al., 2006; Llobet et al., 2008). Shed capsular polysaccharides from A. baumannii appear to form a “binding site barrier” that limits 8E3 access to the bacterial surface, likely contributing to the observed lack of efficacy of 8E3 in the mouse pneumonia model.
The mechanism responsible for the apparent ADE of A. baumannii infection is not completely clear; however, our studies and prior literature reports support two possibilities: 1) soluble 8E3-capsule immune complexes, present in high concentrations in the blood of infected mice after 8E3 dosing, depress the activity of phagocytic cells; and 2) 8E3-A. baumannii complexes enhance infection through increasing adherence to, and invasion of, host cells by interacting with Fc receptors. Although the inhibitory effects of soluble 8E3-K2 complexes on phagocytic cells were not investigated in this work, many published reports clearly demonstrate this phenomenon (Griffin, 1980; Michl et al., 1983; Astry and Jakab, 1984; Halstead et al., 2010). Immune complex–mediated depression of phagocytic activity may lead to reduced A. baumannii clearance in vivo, potentially explaining the observed increases in bacterial load and the observed increases in animal mortality. The second possibility, which has been described for the ADE of viral infection, relates to possible intracellular sanctuaries for A. baumannii (e.g., within epithelial cells) and the enhancement of A. baumannii adhesion and invasion that is mediated by 8E3. We found that the binding of 8E3 to the capsule enhanced adherence/invasion of several carbapenem-resistant K2 clinical isolates to human lung epithelial cells in vitro. Further studies showed that this enhancement of bacterial adherence/invasion was mediated via IgG engagement of FcγRs. 8E3-mediated increases of A. baumannii adherence and/or invasion of lung epithelial cells may result in cellular cytotoxicity over time. Another possible consequence of increased adherence/invasion at high bacterial titers is that some A. baumannii may survive within the epithelial cells, which in turn enhances the survival of these Trojan horse A. baumannii. Although ADE has been primarily described with intracellular pathogens (e.g., viruses) and A. baumannii is primarily an extracellular pathogen, an intracellular niche under certain circumstances is biologically plausible (Choi et al., 2008). Investigations on the ability of A. baumannii to survive within various intracellular environments are needed. These data would assist in delineating the mechanisms that contribute to the observed ADE of A. baumannii infection in mice. Furthermore, adherence-independent mechanisms may also be involved. A human IgM mAb targeting lipid A of Escherichia coli has been shown to increase mortality without altering bacterial load in a canine model of E. coli sepsis (Quezado et al., 1993). The binding of the mAb to shed capsule and hence the formation of antibody-capsule immune complexes may increase inflammatory responses leading to septic shock, which may lead to increased animal mortality.
Antibodies targeting capsular polysaccharides have been previously shown to confer protection against A. baumannii in mouse infection models by Russo et al. (2013) and Nielsen et al. (2017). Although the reasons that the present study results differ from the previous work are not completely clear, there are several possible explanations. First, the effects of mAb treatment may be highly dependent on the infection model that is used. It is possible that anti-capsule mAbs may provide protection in animal models with relatively low bacterial inocula (e.g., ≤1 × 108 CFU), and with relatively low blood concentrations of shed capsule. Nielsen et al. (2017) recently developed an anti-capsule IgG1 mAb C8 and showed protection against XDR A. baumannii in mouse sepsis and pneumonia models inoculated with 107–108 CFU bacteria (Nielsen et al., 2017). In contrast, in conditions of high bacterial loads (e.g., as in the mouse pneumonia model with a bacterial inoculum of 109 CFU), high concentrations of 8E3-capsule complexes may suppress phagocytic bacterial killing. Of note, significantly diminished in vivo efficacy of mAb C8 was observed when administered ≥1 hour postinfection (Nielsen et al., 2017), consistent with a prophylactic effect, possibly due to lower bacterial loads and proinflammatory responses at the beginning of infection compared with antibody treatment at later stages of infection when the bacterial burden is increased. Different routes of antibody administration may also contribute to the differences, as the work of Nielsen et al. (2017) employed intravenous dosing and our work employed intraperitoneal dosing (delaying the attainment of peak concentrations of mAb in blood). Second, antibody isotype may impact efficacy as evidenced by the observations that both anti-capsule IgG1 and IgM provided protection against A. baumannii in animal infection models (Russo et al., 2013; Nielsen et al., 2017). mIgG3 is an early effector molecule that can be produced by B cells independently of T-cell help, and hence it plays an important role against T cell–independent antigens such as bacterial polysaccharides (Harmer and Chahwan, 2016). ADE of A. baumannii infection may be IgG3 isotype specific because of the role of bacterial adherence and colonization in the initiation of infection. Third, the capsule type of A. baumannii may also affect the result. Capsule types K4 (strain HUMC1) and K1 (strain AB307-0294) were used in previous studies, but capsule type K2 (strain AB899) was used in the present work. It is unclear whether capsule type–specific virulence factors contributed to the observed ADE of infection in mice.
In summary, we developed an mIgG3 mAb directed against the K2 capsular polysaccharide of A. baumannii. Opsonization with 8E3 resulted in concentration-dependent phagocytic killing of AB899 by murine macrophages. However, passive immunization with 8E3 led to enhanced AB899 bacterial growth and increased mortality in a murine pneumonia model. A rapid decrease of plasma-free 8E3 concentration was observed in A. baumannii–infected mice, which is likely due to the large binding capacities of AB899 and shed capsule components. These results raise concerns regarding the suitability of capsular polysaccharides as therapeutic targets for passive immunization, as shed capsule may mediate a “decoy effect,” which may serve as a barrier to effective engagement of capsule on bacterial cells in infected tissues and mAb-shed capsule immune complexes may also depress bacterial clearance by phagocytic cells. The potential of antibodies to enhance the infection of A. baumannii raises concerns for antibody-based therapy. Future studies examining the effects of antibody isotypes, target epitopes, bacterial capsule types, intracellular A. baumannii, capsule-antibody complexes, animal models, and prophylactic versus postinfection treatment will help to better understand the mechanism of ADE of A. baumannii infection.
Acknowledgments
We thank Nicole R. Luke-Marshall for assistance in antibody development and Maureen Adolf for help in animal studies.
Authorship Contributions
Participated in research design: Wang-Lin, Russo, Balthasar.
Conducted experiments: Wang-Lin, Olson, Beanan, MacDonald, Russo.
Performed data analysis: Wang-Lin.
Wrote or contributed to the writing of the manuscript: Wang-Lin, Russo, Balthasar.
Footnotes
- Received September 13, 2018.
- Accepted January 2, 2019.
This work was supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (Grant IBX000984A) (T.A.R.); and through funding provided by the Center of Protein Therapeutics (J.P.B.).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AB
- capsule type in Acinetobacter baumannii
- ADE
- antibody-dependent enhancement
- ATCC
- American Type Culture Collection
- AUC
- area under the concentration-time curve
- AUC0–24 h
- AUC for 0–24 hours
- CFU
- colony-forming units
- dA/dt
- change of absorbance over time
- ELISA
- enzyme-linked immunosorbent assay
- EU
- endotoxin units
- FcγR
- Fc gamma receptor
- FITC
- fluorescein isothiocyanate
- Kd
- equilibrium dissociation constant
- LB
- lysogeny broth
- LDH
- lactate dehydrogenase
- LLOD
- lower limit of detection
- LLOQ
- lower limit of quantification
- mAb
- monoclonal antibody
- mIgG3
- mouse IgG3
- MOI
- multiplicity of infection
- PBS
- phosphate-buffered saline
- QC
- quality control sample
- t1/2
- terminal half-life
- TCA
- trichloroacetic acid
- XDR
- extensively drug resistant
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}