


Visual Overview
Abstract
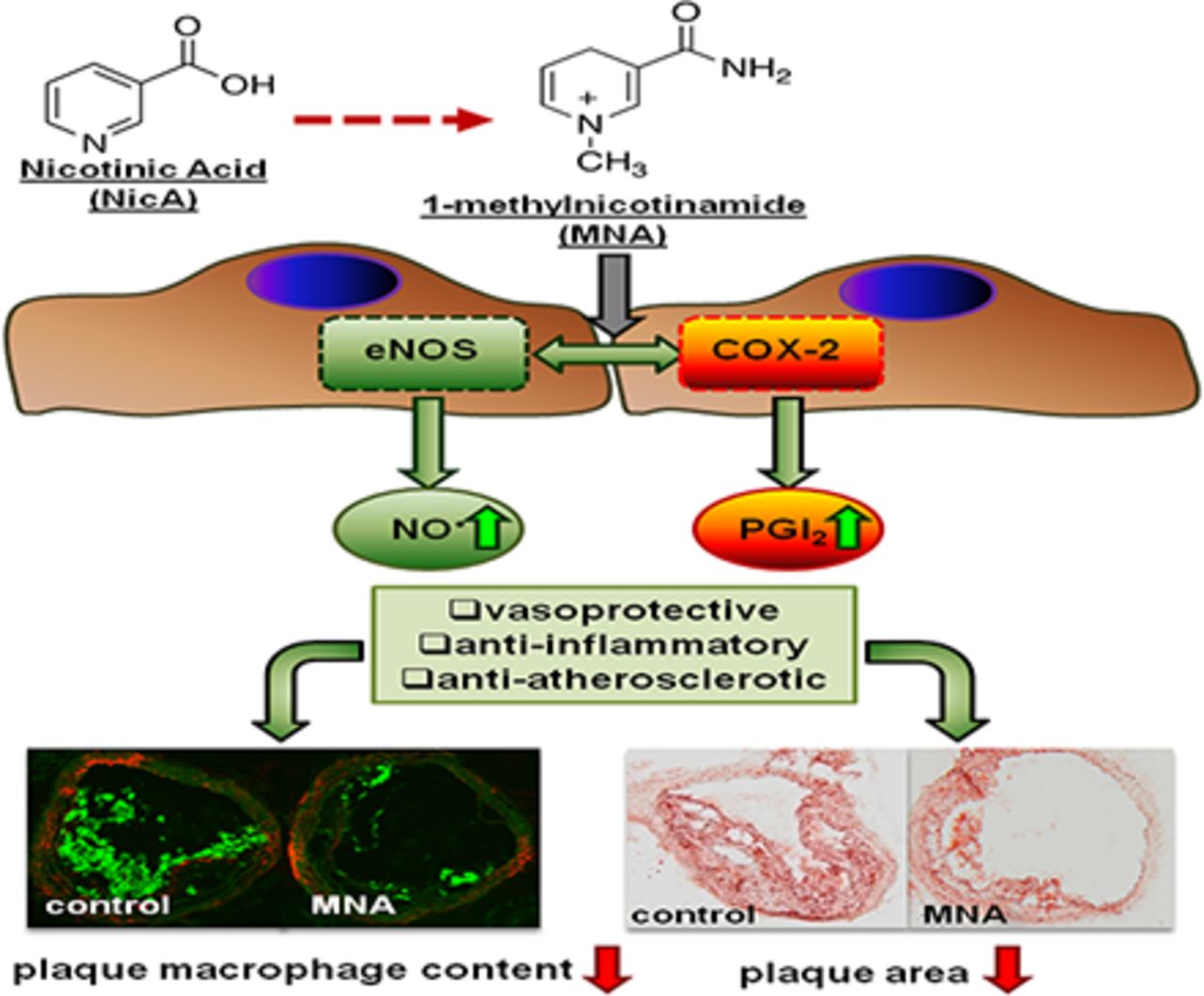
1-Methylnicotinamide (MNA), the major endogenous metabolite of nicotinic acid (NicA), may partially contribute to the vasoprotective properties of NicA. Here we compared the antiatherosclerotic effects of MNA and NicA in apolipoprotein E (ApoE)/low-density lipoprotein receptor (LDLR)–deficient mice. ApoE/LDLR−/− mice were treated with MNA or NicA (100 mg/kg). Plaque size, macrophages, and cholesterol content in the brachiocephalic artery, endothelial function in the aorta, systemic inflammation, platelet activation, as well as the concentration of MNA and its metabolites in plasma and urine were measured. MNA and NicA reduced atherosclerotic plaque area, plaque inflammation, and cholesterol content in the brachiocephalic artery. The antiatherosclerotic actions of MNA and NicA were associated with improved endothelial function, as evidenced by a higher concentration of 6-keto-prostaglandin F1α and nitrite/nitrate in the aortic ring effluent, inhibition of platelets (blunted thromboxane B2 generation), and inhibition of systemic inflammation (lower plasma concentration of serum amyloid P, haptoglobin). NicA treatment resulted in an approximately 2-fold higher concentration of MNA and its metabolites in urine and a 4-fold higher nicotinamide/MNA ratio in plasma, compared with MNA treatment. In summary; MNA displays pronounced antiatherosclerotic action in ApoE/LDLR−/− mice, an effect associated with an improvement in prostacyclin– and nitric oxide–dependent endothelial function, inhibition of platelet activation, inhibition of inflammatory burden in plaques, and diminished systemic inflammation. Despite substantially higher MNA availability after NicA treatment, compared with an equivalent dose of MNA, the antiatherosclerotic effect of NicA was not stronger. We suggest that detrimental effects of NicA or its metabolites other than MNA may limit beneficial effects of NicA-derived MNA.
Footnotes
- Received August 27, 2015.
- Accepted November 30, 2015.
This research was supported by the European Union from the resources of the European Regional Development Fund under the Innovative Economy Programme [Grant POIG.01.01.02-00-069/09]. S.C. is a coinventor of patent no. CA2547234 A1 (“The use of quaternary pyridinium salts as vasoprotective agents”).
We would like to thank Dr. Jan Adamus from Lodz University of Technology for kindly providing us with MNA, and Dr. Christoph Johann from Wyatt Tech. for FFF method-based measurements of lipid profile.
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}