


Abstract
The present study used human myeloid leukemia U937 cells, a versatile promonocytic cellular system that, based on its endoplasmic reticulum (ER)/mitochondria functional relationships, responds to low micromolar concentrations of arsenite with a single, defined mechanism of superoxide (O2−.) formation. Under these conditions, we observe an initial Ca2+ mobilization from the ER associated with the mitochondrial accumulation of the cation, which is followed by Ca2+-dependent mitochondrial O2−. (mitoO2−.) formation. These events, which were barely detectable after 3 hours, were better appreciated at 6 hours. We found that markedly shorter exposure to arsenite and lower concentrations of arsenite are required to induce extensive O2− formation in cells supplemented with inositol-1,4,5-trisphosphate receptor (IP3R) or ryanodine receptor (RyR) agonists. Indeed, nanomolar arsenite induced maximal O2−. formation after only 10 minutes of exposure, and this response was uniquely dependent on the enforced mitochondrial Ca2+ accumulation. The dramatic anticipation of and sensitization to the effects of arsenite caused by the IP3R or RyR agonists were accompanied by a parallel significant genotoxic response in the absence of detectable mitochondrial dysfunction and cytotoxicity. We conclude that the prolonged, low-micromolar arsenite exposure paradigm resulting in mitoO2−. formation is necessary to affect Ca2+ homeostasis and accumulate the cation in mitochondria. The arsenite requirements to promote mitoO2−. formation in the presence of sufficient mitochondrial Ca2+ were instead remarkably lower in terms of both concentration and time of exposure. These conditions were associated with the induction of extensive DNA strand scission in the absence of detectable signs of toxicity.
SIGNIFICANCE STATEMENT In respiration-proficient cells, arsenite causes mitochondrial Ca2+ accumulation and Ca2+-dependent mitochondrial superoxide formation. We now report that the second event requires remarkably lower concentrations of and time of exposure to the metalloid than the former. Indeed, a brief exposure to nanomolar levels of arsenite produced maximal effects under conditions in which the mitochondrial Ca2+ concentration ([Ca2+]m) was increased by inositol-1,4,5-trisphosphate receptor or ryanodine receptor agonists. Hence, specific substances or conditions enhancing the [Ca2+]m may potentiate the deleterious effects of arsenite by selectively increasing mitochondrial superoxide formation.
Introduction
Arsenite is a well established human carcinogen (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2004) that, because of its ubiquitous presence in the environment, represents a serious threat for millions of people (Shakoor et al., 2017). Indeed, ingestion of contaminated drinking water by populations from various parts of the planet has been associated with the induction of different types of tumors [lung, skin, liver, bladder, and kidney (Sankpal et al., 2012; Shakoor et al., 2017; Zhou and Xi, 2018)] as well as other nonmalignant diseases affecting various tissues and organs (Flora, 2011; Shakoor et al., 2017).
Arsenite therefore mediates a wide spectrum of deleterious effects, bearing numerous consequences in different cells and organs, a complex scenario in apparent contrast with the simple mechanism often implicated in the effects of the metalloid. More specifically, the opinion of various groups is that a large proportion of the critical effects mediated by arsenite result from the intermediate formation of reactive oxygen species (ROS) (Flora, 2011; Minatel et al., 2018).
Hence, the impact of the metalloid in different cell types should depend on their susceptibility to arsenite-induced ROS formation, which is in turn affected by an array of variables associated with the specific characteristics of the cells, tissues, and organs. Different cells respond to arsenite by producing ROS via different mechanisms, e.g., in the mitochondrial respiratory chain (Liu et al., 2005; Guidarelli et al., 2015, 2016a), via NADPH oxidase activation (Smith et al., 2001; Chou et al., 2004; Straub et al., 2008), or through both mechanisms (Li et al., 2014).
Mechanistic studies on arsenite cytotoxicity and genotoxicity should therefore ideally involve the use of specific cell types that are characterized by a well defined antioxidant system and respond to arsenite through established mechanisms of ROS formation. In this direction, we performed our initial studies on arsenite toxicity in U937 cells, from now on referred to as respiration-proficient cells (RP cells), demonstrating that these cells uniquely produce mitochondrial superoxide (mitoO2−.) in response to a low concentration of arsenite (Guidarelli et al., 2015, 2016a). This ROS response was sensitive to inhibitors of electron transport in the respiratory chain as well as to the respiration-deficient phenotype, i.e., respiration-deficient cells (RD cells) derived from the same RP cell line, which failed to produce mitoO2−. in response to arsenite. We then investigated the mechanism whereby arsenite mobilizes Ca2+ from the endoplasmic reticulum (ER) and found that the metalloid promotes an initial slow mobilization of a limited amount of the cation from the inositol-1,4,5-trisphosphate receptor (IP3R). This response was ROS-independent and was mediated by the direct interaction of arsenite with this Ca2+ pool (Guidarelli et al., 2018). The initial IP3R stimulation was associated with additional direct effects of the metalloid on the intraluminal cross talk between the IP3R and the ryanodine (Ry) receptor (RyR), thereby leading to the release of large amounts of Ca2+ from the RyR (Guidarelli et al., 2018). This second response was therefore sensitive to RyR inhibitors and RyR downregulation (Guidarelli et al., 2009).
Finally, the fraction of Ca2+ derived from the RyR was taken up by the mitochondria through the mitochondrial Ca2+ uniporter (MCU), in which the cation critically contributed to events promoting mitoO2−. formation (Guidarelli et al., 2019a); that is, mitoO2−. formation induced by arsenite in RP cells, besides being sensitive to inhibitors of electron transport and the respiration-deficient phenotype, was also suppressed by IP3R, RyR, and MCU inhibitors. In addition, the metalloid failed to promote mitoO2−. formation in cells with downregulated RyR (Guidarelli et al., 2019a).
The mechanisms mediating ROS formation in RP cells exposed to arsenite therefore involve upstream events directly induced by the metalloid through its binding in specific sites of the ER, leading to Ca2+ mobilization and mitochondrial accumulation (Guidarelli et al., 2019a). The slow effects on the ER are possibly dependent on the time necessary to allow a significant cellular uptake of the metalloid and promote effects resulting in Ca2+ mobilization and mitochondrial accumulation. It is, however, unclear whether the same prolonged exposure to specific arsenite concentrations is also necessary to trigger the Ca2+-dependent mitochondrial events associated with superoxide (O2−.) formation.
These considerations stimulated the present study, in which we investigated the requirements for arsenite in the overall process leading to mitochondrial O2−. formation. More specifically, we asked the question of whether IP3R or RyR agonist–induced enforced mitochondrial Ca2+ accumulation shortens the time required by and/or lowers the concentration of arsenite to promote the formation of mitoO2−..
Materials and Methods
Chemicals.
Sodium arsenite, 2-aminoethoxydiphenyl borate (2-APB), Ry, L-ascorbic acid (AA), rotenone, caffeine (Cf), ATP, phorbol 12-myristate 13-acetate (PMA), apocynin, diphenyleneiodonium (DPI), Hoechst 33342, and most of the reagent-grade chemicals were purchased from Sigma-Aldrich (Milan, Italy). Ru360 was from Thermo Fisher Scientific (Milan, Italy). Fluo-4- acetoxymethyl ester (AM), Rhod 2-AM, dihydrorhodamine 123 (DHR), and MitoSOX Red were purchased from Thermo Fisher Scientific.
Antibodies.
The antibodies against the phosphorylated or non phosporylated p47 phagocyte oxidase (p47phox) were from Sigma-Aldrich. The antibody against thioredoxin-2 (Trx2; sc-133201) and the horseradish peroxidase–conjugated secondary antibody were from Santa Cruz (Santa Cruz, CA).
Cell Culture and Treatment Conditions.
U937 human myeloid leukemia cells, herein defined as RP cells, were cultured in suspension in RPMI 1640 medium (Sigma-Aldrich). Culture media were supplemented with 10% FBS (Euroclone, Celbio Biotecnologie, Milan, Italy), penicillin (100 U/ml), and streptomycin (100 mg/ml) (Euroclone). Cells were grown at 37°C in T-75 tissue culture flasks (Corning Inc., Corning, NY) gassed with an atmosphere of 95% air and 5% CO2. The respiration-deficient phenotype (RD cells) was induced as indicated in Guidarelli et al. (2016b).
Sodium arsenite was prepared as a 1 mM stock solution in saline A (8.182 g/l NaCl, 0.372 g/l KCl, 0.336 g/l NaHCO3, and 0.9 g/l glucose, pH 7.4) and stored at 4°C. Cells (1 × 105 cells/ml) were exposed to arsenite and/or other additions in complete RPMI 1640 culture medium, as detailed in the text and figure legends. Treatments with AA were performed as detailed elsewhere (Guidarelli et al., 2014).
DHR and MitoSOX Red Fluorescence Assay.
The cells were supplemented with either 10 µM DHR or 5 µM MitoSOX Red for 30 minutes and subsequently exposed for the last 10 minutes to arsenite. In other experiments, cells were exposed for increasing time intervals to arsenite, and DHR was added to the culture medium in the last 30 minutes of incubation. After the treatments, the cells were washed three times with saline A, and fluorescence images were captured with a BX-51 microscope (Olympus, Milan, Italy) equipped with a SPOT-RT camera unit (Diagnostic Instruments, Delta Sistemi, Rome, Italy) using an Olympus LCAch 40×/0.55 objective lens. The excitation and emission wavelengths were 488 and 515 nm (DHR) and 510 and 580 nm (MitoSOX red), with a 5-nm slit width for both emission and excitation. Images were collected with exposure times of 100–400 milliseconds, digitally acquired, and processed for fluorescence determination at the single-cell level on a personal computer using ImageJ software. Mean fluorescence values were determined by averaging the fluorescence values of at least 50 cells per treatment condition per experiment.
Measurement of Intracellular Free Calcium Levels and Mitochondrial Ca2+.
Cells were treated for 30 minutes with either 4 µM Fluo-4-acetoxymethyl ester or 10 µM Rhod 2-acetoxymethyl ester and subsequently exposed for the last 10 minutes to arsenite. After the treatments, the cells were washed three times with saline A and subsequently analyzed with a fluorescence microscope. The resulting images were taken and processed as described above. The excitation and emission wavelengths were 488 and 515 nm (Fluo-4) and 540 and 590 nm (Rhod-2), with a 5-nm slit width for both emission and excitation. Images were collected with exposure times of 100–400 milliseconds, digitally acquired, and processed for fluorescence determination at the single-cell level on a personal computer using the ImageJ software. Mean fluorescence values were determined by averaging the fluorescence values of at least 50 cells per treatment condition per experiment.
Western Blot Analysis.
After treatments, the cells were lysed as described in Guidarelli et al. (2019a). Equal amounts of proteins (25 µg) were loaded in each lane, separated by PAGE in the presence of SDS, transferred to polyvinyldiene difluoride membranes, and probed with antibodies against phosphorylated p47phox or p47phox. Details on Western blotting apparatus and conditions are reported elsewhere (Guidarelli et al., 2019a). Antibody against p47phox was used to assess the equal loading of the lanes.
Redox Western Blot Analysis.
The Trx2 redox state was estimated by redox Western blots as described in Folda et al. (2016). Briefly, after treatments, the cells (2.5 × 106) were washed with PBS (136 mM NaCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, 3 mM KCl; pH 7.4) and resuspended in 150 µl of urea lysis buffer (100 mM Tris/HCl, pH 8.2; urea 8 M; EDTA 1 mM) containing 10 mM iodoacetoamide. The samples were then incubated for 20 minutes at 37°C and centrifuged at 14,000g for 1 minute. Ten volumes of cold acetone/1 M HCl (98:2) were added to the supernatants, and the pellets were washed twice with acetone/1 M HCl/H2O (98:2:10). The pellets were resuspended in 60 µl of urea lysis buffer containing 3.5 mM dithiothreitol and, after a 30-minute incubation at 37°C, supplemented with 30 mM iodoacetoamide and incubated for a further 30 minutes at 37°C. Samples were then subjected to urea-PAGE (7 M urea and 7% acrylamide) under nonreducing conditions and blotted (Folda et al., 2016).
Fluorogenic Caspase 3 Assay.
Caspase 3–like activity was monitored as described in Fiorani et al. (2018). Briefly, the cells were lysed, and aliquots of the extract (30 μg proteins) were incubated with 12 μM of Acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin, at 30°C. Caspase 3–like activity was determined fluorometrically (excitation at 360 nm and emission at 460 nm) by quantifying the release of aminomethylcoumarin from cleaved caspase 3 substrate at appropriate intervals.
Analysis of Apoptosis with Hoechst 33342 Assay.
After treatments, the cells were incubated for 5 minutes in the presence of 10 μM Hoechst 33342 and then analyzed with a fluorescence microscope to assess their nuclear morphology (chromatin condensation and fragmentation). Cells with homogeneously stained nuclei were considered viable.
Cytotoxicity Assay.
After treatments with arsenite, the number of viable cells was estimated with the trypan blue exclusion assay. Briefly, an aliquot of the cell suspension was diluted 1:2 (v/v) with 0.4% trypan blue, and the viable cells (i.e., those excluding trypan blue) were counted with a hemocytometer.
Alkaline Halo Assay.
DNA single-strand breakage was determined using the alkaline halo assay that was developed in our laboratory (Crimella et al., 2011). It is important to note that, although we refer to DNA strand scission throughout the text, the DNA nicks measured by this technique under alkaline conditions may in fact include alkali labile sites in addition to direct strand breaks. Details on the alkaline halo assay, processing of fluorescence images, and the calculation of the experimental results are also given in Crimella et al., 2011. DNA single-strand breakage was quantified by calculating the nuclear spreading factor value, which represents the ratio between the area of the halo (obtained by subtracting the area of the nucleus from the total area, nucleus + halo) and that of the nucleus, from 50 to 75 randomly selected cells per experiment per treatment condition.
ATP Determination.
Ice-cold 5% perchloric acid was added to the cells (2 × 106 cells). After a 10-minute incubation in an ice bath, the samples were centrifuged for 5 minutes at 10,000g. The supernatants were neutralized with 3 M K2CO3, and the precipitates were removed by centrifugation. Ten percent (v/v) 1 M KH2PO4 (pH 6.5) was added to the nucleotide-containing supernatants. The samples were then filtered through 0.22-μm pore microfilters. The clear filtered solutions were directly analyzed by high-performance liquid chromatography (HPLC) with the UV detection wavelength set at 254 nm (Stocchi et al., 1985). The liquid chromatographic system used consisted of a System Gold Programmable Solvent Module 125 and a System Gold Programmable Solvent Detector 166 (Beckman, Fullerton, CA). The HPLC separation was performed using a 5-µm Supelco Discovery C18 column (15 cm × 4.6 mm) equipped with a 5-µm Supelguard Discovery 18 (2 cm × 4.0 mm) (Supelco, Bellefonte, PA). The mobile phase used for the separation consisted of buffer A (0.1 M KH2PO4) and buffer B (90:10 buffer A/methanol). The following gradient of buffer B was used: 25% in 9 minutes, 90% in 6 minutes, and 100% in 2.5 minutes. Buffer B (100%) was held for 2 minutes, and 100%–0% was reached in 6 minutes. Re-equilibration with buffer A for 5 minutes was performed before each injection. The injected volume was 20 µl, and the flow rate was 1.3 ml/min.
Measurement of GSH Content by High-Performance Liquid Chromatography.
The cells (1 × 106) were suspended in 100 µl of lysis buffer, vortexed, and kept for 10 minutes on an ice bath. Thereafter, 15 µl of 0.1 N HCl and 140 µl of precipitating solution (0.2 M glacial meta-Phosphoric acid, 5 mM sodium EDTA, 5 M NaCl) were added to the samples. After centrifugation, the supernatants were collected and kept at −20°C until the HPLC analyses. Just before analysis, 60 µl of the acid extract was supplemented with 15 µl of 0.3 M Na2HPO4 and 15 µl of a solution containing 20 mg of 5,5'-dithiobis(2-nitrobenzoic acid) in 100 ml of sodium citrate (1% w/v). The mixture was stirred for 1 minute at room temperature and, after 5 minutes, filtered through 0.22-µm pore microfilters. The clear filtered solution was directly analyzed by HPLC, with the UV detection wavelength set at 330 nm for their glutathione (GSH) content (Brundu et al., 2016), using a 15 cm × 4.6 mm, 5-µm Supelco Discovery C18 column (Supelco, Bellefonte, PA). The UV absorption was detected at 330 nm. The injection volume was 20 µl. The retention time of GSH was approximately 15.7 minutes.
The liquid chromatographic system used is the same described in the previous paragraph (ATP determination). The HPLC separation was performed using a 15 cm × 4.6 mm, 5-µm Supelco Discovery C18 column equipped with a 5-µm Supelguard Discovery 18 (2 cm × 4.0 mm). The mobile phase used for the separation consisted of buffer A (0.1 M KH2PO4) and buffer B (40:60 buffer A/acetonitrile). The following gradient of buffer B was used: 0%–100% in 15 minutes; 100% buffer B was held for 5 minutes, and 100%–0% was reached in 20 minutes. Re-equilibration with buffer A for 10 minutes was performed before each injection. The injected volume was 20 µl, and the flow rate was 1 ml/min.
Statistical Analysis.
The results are expressed as means ± S.D. Statistical differences were analyzed by one-way ANOVA followed by Dunnett’s test for multiple comparison or two-way ANOVA followed by Bonferroni’s test for multiple comparison using Prism 6.0 software (GraphPad Software). A value of P < 0.05 was considered significant.
Results
RyR or IP3R Agonists Burst ROS Formation after a Very Short Time of Exposure to Very Low Concentrations of Arsenite.
In keeping with our recent findings (Fiorani et al., 2018), exposure of RP cells to 2.5 µM arsenite leads to a very slow, time-dependent DHR fluorescence response that is barely detectable after 3 hours, more clearly appreciable at 6 hours, and even more significant at 16 hours (Fig. 1A). It was then interesting to observe that this time dependence is lost under conditions in which the cells also receive IP3R or RyR agonists in the last 10 minutes of arsenite exposure. More specifically, we found that 100 μM ATP, an agonist of metabotropic purinergic receptors causing IP3R activation (Berridge, 1993), or 10 mM Cf (a RyR agonist) (Meissner, 2017) causes a remarkable leftward shift in the time dependence of the DHR fluorescence response mediated by the metalloid. Indeed, the responses detected in cells exposed to arsenite alone for 16 hours were identical to those observed 1 or 2 hours after addition of the metalloid under conditions of ATP or Cf supplementation in the final 10 minutes. To determine the effects mediated by shorter exposure to arsenite, we changed the above experimental protocol and loaded the cells with DHR prior to arsenite exposure. Interestingly, the fluorescence responses detected after a 30-minute treatment with arsenite, with ATP or Cf added in the last 10 minutes, or even under conditions of concomitant 10-minute exposure to the metalloid and ATP or Cf (Fig. 1B) were identical to those described above and illustrated in Fig. 1A. The concentrations of ATP and Cf were selected on the bases of previous concentration dependence studies, which show that under these conditions both agents produce maximal increases in the cytosolic ([Ca2+]c) and mitochondrial ([Ca2+]m) Ca2+ concentrations (Guidarelli et al., 2018). The enhancing effects of ATP and Cf on the arsenite-induce DHR fluorescence response displayed the same concentration dependence (data not shown).

Time and concentration dependence for the arsenite-induced ROS formation in the absence or presence of Ca2+ mobilizing agents. (A) RP cells were exposed for increasing time intervals to 2.5 μM arsenite, supplemented in the last 30 and 10 minutes of incubation with DHR and ATP (100 μM) or Cf (10 mM), respectively, and finally analyzed for the resulting DHR fluorescence responses. (B) Cells were preloaded with DHR (30 minutes), treated for 10 or 30 minutes with arsenite, and exposed to ATP or Cf in the last 10 minutes of incubation. (C) Cells were preloaded with DHR (30 minutes) and then exposed for 10 minutes to ATP or Cf with or without increasing concentrations of arsenite. (D) Cells preloaded with DHR (30 minutes) were exposed for 5 minutes to the vehicle, 2-APB (50 μM), Ry (20 μM), or Ru360 (10 μM) and finally treated for a further 10 minutes with 2.5 μM arsenite with or without ATP or Cf. After treatments, the cells were analyzed for their DHR fluorescence. Results represent the means ± S.D. calculated from at least three separate experiments. *P < 0.05; **P < 0.01, as compared with untreated cells. (A–C) Two-way ANOVA followed by Bonferroni’s test; (D) one-way ANOVA followed by Dunnet’s test.
The next question addressed was on the arsenite concentration dependence. For this purpose, the cells were treated for 10 minutes with increasing concentrations of arsenite associated with either ATP or Cf. The results obtained in these experiments are shown in Fig. 1C and indicate that ROS formation is not further increased at arsenite concentrations greater than 2.5 µM, which was somewhat expected based on the above results. These results indeed demonstrated identical ROS· emission after very short- or long-term incubation with arsenite, which presumably should be associated with significantly different cellular accumulation of the metalloid. Much less expected was the outcome of experiments using arsenite concentrations in the submicromolar range. We found that the same maximal response detected with 2.5 µM arsenite was obtained using a 5-fold lower concentration of the metalloid and that 100 nM arsenite was sufficient to induce a statistically significant effect.
To correctly interpret the significance of the above findings, it is important to keep in mind that the fluorescence probe employed in the ROS experiments, DHR, allows the detection of H2O2 and various types of ROS, including the O2−. and the hydroxyl radical (Gomes et al., 2005). DHR also detects reactive N species as peroxynitrite, which cannot be produced in the specific cell line employed (Guidarelli et al., 2016a). Our previous studies (Guidarelli et al., 2015, 2016a, 2017), as summarized in the Introduction section, demonstrated that the slow ROS response induced by arsenite is uniquely indicative of mitoO2−. formation, which then dismutates to diffusible H2O2, causing the oxidation of DHR in the cytosolic compartment. It is instead to be determined whether the IP3R or RyR agonists promote an enhanced DHR fluorescence response by potentiating the same mechanisms or by recruiting different mechanisms, such as NADPH oxidase.
We therefore conclude that a very short exposure to very low concentrations of arsenite promotes maximal ROS formation in the presence of ATP or Cf. The notion that these enhancing responses do not simply appear maximal because of a limitation of the assay was established by showing that increasing concentrations of H2O2 promote a linear fluorescence response up to values two to three times greater than those obtained in the above experiments (data not shown).
The Enhancing Effects of the RyR or IP3R Agonists on ROS Formation are Linked to the Enforced Mitochondrial Ca2+ Accumulation.
We performed experiments to determine the mechanism of ROS formation induced by arsenite in cells supplemented with ATP or Cf. In recent studies, we investigated the effects of these agonists on RP cell Ca2+ homeostasis (Guidarelli et al., 2018, 2019a) and therefore decided to recapitulate some of these findings to finally determine whether these responses are affected by arsenite. In other words, we tested the effect of the short-term coexposure paradigms on Ca2+ homeostasis.
We found that arsenite alone fails to promote detectable effects in the [Ca2+]c (Supplemental Fig. 1A) and [Ca2+]m (Supplemental Fig. 1B), as expected. Cf instead promoted a weak increase in the [Ca2+]c (Supplemental Fig. 1A), which was associated with a robust increase in [Ca2+]m (Supplemental Fig. 1B). These responses were not affected by arsenite and, under these conditions, were insensitive to the IP3R antagonist 2-APB (Maruyama et al., 1997) and prevented by Ry, an RyR antagonist (Meissner, 2017). Furthermore, the MCU inhibitor Ru360 (Zazueta et al., 1999) selectively abolished the increase in the [Ca2+]m, with hardly any effect detected on the [Ca2+]c.
We next found that ROS formation, determined by monitoring DHR oxidation, was not induced by arsenite alone but was readily detected after combined exposure to the metalloid and Cf. In addition, as observed for the increase in the [Ca2+]m, this response was sensitive to Ry and Ru360, with hardly any effect detected with 2-APB (Fig. 1D).
In other experiments, we used ATP in place of Cf to induce a significantly greater increase in the [Ca2+]c (Supplemental Fig. 1A), associated with the same increase in [Ca2+]m previously detected with the RyR agonist (Supplemental Fig. 1B). These effects, which were mediated by ATP, were not affected by arsenite and, under these conditions, were prevented by 2-APB. In addition, Ry was as effective as Ru360 in preventing the increase in [Ca2+]m, with only minor (Ry) or no (Ru360) effects being detected on the [Ca2+]c. ROS formation was observed after combined exposure to ATP and arsenite and was sensitive to the above treatments preventing mitochondrial Ca2+ accumulation (Fig. 1D). Finally, ATP or Cf failed to promote ROS formation in the absence of arsenite.
Collectively, the above results indicate that the mechanism whereby the IP3R and RyR agonists enhance ROS formation induced by a 10-minute exposure to arsenite involves enforced mitochondrial Ca2+ accumulation. The experimental approach employed in the above experiments also provided evidence for the specificity of the effects mediated by the antagonists and inhibitors.
Short-Term Exposure to Arsenite in Combination with a RyR or IP3R Agonist Uniquely Mediates mitoO2−. Formation.
We asked the question of whether the enhancing effects mediated by IP3R or RyR agonists are associated with an increased mitoO2−. formation or, rather, to the recruitment of a different mechanism, such as NADPH activation.
As indicated in Fig. 2A, the DHR fluorescence responses mediated by the short-term exposure to arsenite in combination with either Cf or ATP are prevented by the complex I inhibitor rotenone, as well as by AA, supplemented under conditions promoting high and low concentrations of the vitamin in the mitochondria and cytosol, respectively (Fiorani et al., 2015; Cantoni et al., 2018). In addition, rotenone or AA failed to affect the changes in the [Ca2+]m elicited by Cf or ATP in combination with the metalloid (Fig. 2B). Most importantly, the effects mediated by rotenone were reproduced by the respiration-deficient phenotype, i.e., RD cells failed to respond to arsenite/ATP or arsenite/Cf with an increased DHR oxidation (Fig. 2A) but nevertheless responded to these treatments with increases in [Ca2+]m comparable with those detected in their respiration-proficient counterparts (Fig. 2B).

ROS formation induced by short-term coexposure to low concentrations of arsenite and the Ca2+ mobilizing agents takes place in the mitochondrial respiratory chain. (A–C) RP and RD cells preloaded for 30 minutes with DHR (A), Rhod-2-AM (B), or MitoSOX red (C) were exposed for 5 minutes to the vehicle, rotenone (0.5 μM), or AA (10 μM) and were finally treated for a further 10 minutes with 2.5 μM arsenite with or without ATP or Cf. After treatments, the cells were analyzed for the resulting fluorescence responses. Results represent the means ± S.D. calculated from at least three separate experiments. *P < 0.01, as compared with untreated cells (one-way ANOVA followed by Dunnet’s test).
The mitochondrial origin of ROS produced using the above treatment paradigms was next confirmed using MitoSOX red, a fluorochrome commonly employed for the detection of O2−. in the mitochondria of live cells (Mukhopadhyay et al., 2007). Indeed, as indicated in Fig. 2C, the results obtained using MitoSOX red were in line with those previously discussed for experiments using DHR. The notion that MitoSOX red, unlike DHR, only detects mitochondrial ROS was previously established by showing the exclusive sensitivity of the second probe to ROS generated via PMA-induced NADPH oxidase activation (Guidarelli et al., 2016b). These experiments, which are not reported here for the sake of brevity, were run in parallel with those described above as internal controls.
We next tested the possibility that the increase in [Ca2+]c mediated by the 10-minute exposure paradigm was associated with an early activation of NADPH oxidase, which at least in principle may eventually mediate downstream stimulation of mitochondrial ROS formation. For this purpose, we performed experiments using ATP, which causes a greater increase in [Ca2+]c than Cf (Supplemental Fig. 1A). The results illustrated in Supplemental Fig. 2A indicate that the PMA-induced DHR fluorescence response, unlike the one induced by a 10-minute exposure to ATP/arsenite, was sensitive to the NADPH oxidase inhibitors apocynin (10 μM) and DPI (1 μM) (Brandes et al., 2014). Furthermore, PMA also promoted an apocynin- or DPI-sensitive p47phox phosphorylation, instead, which was not detected in cells exposed for 10 minutes to ATP/arsenite (Supplemental Fig. 2B).
In conclusion, the short-term exposure to arsenite in combination with a RyR or IP3R agonist leads to the exclusive formation of mitoO2−., thereby implying that the ROS response induced by arsenite alone is slow for the simple reason that the Ca2+ response leading to increased [Ca2+]m is also slow.
The Short-Term Exposure to Arsenite in Combination with an RyR or IP3R Agonist Fails to Induce Immediate or Delayed Toxicity but Nevertheless Causes Significant DNA Strand Scission.
We initially tested whether the fast mechanism of mitoO2−. formation triggered through the enforced mitochondrial Ca2+ accumulation causes immediate consequences on the thiol pool and energetic status of the cells. We found that a 10-minute exposure to arsenite, Cf, or the two agents combined fails to affect the levels of GSH (untreated cells: 35.06 ± 4.2 nmol/mg proteins) and ATP (untreated cells: 21.05 ± 2.2 nmol/mg proteins). Significant effects were instead detected using H2O2 as a positive control (data not shown). In as much as Trx2 represents a critical controller of ROS emission in mitochondria (Stanley et al., 2011; Folda et al., 2016), we also tested the impact of the cocktail arsenite/Cf or arsenite/ATP on the redox state of this protein. The redox Western blot technique (Supplemental Fig. 3) failed to provide evidence of Trx2 oxidation in cells exposed to arsenite alone or combined with either Cf or ATP. In contrast, the thiol specific oxidant diamide induced a concentration-dependent oxidation of Trx2, with bands appearing in the lower part of the gel.
In other experiments, the cells were treated for 10 minutes with 2.5 μM arsenite alone or associated with ATP or Cf and were subsequently analyzed after 0, 6, or 48 hours of growth in drug-free medium. Under all these different conditions, we found no evidence for caspase 3 activation (Fig. 3A) and chromatin condensation or fragmentation (Fig. 3B). In addition, the cells receiving the different treatments proliferated with rates comparable with those of the untreated cell population (Fig. 3C). Continuous exposure for 48 hours to 2.5 μM arsenite, as we previously reported (Guidarelli et al., 2015, 2016a, 2017), instead caused caspase 3 activation (Fig. 3A), apoptotic DNA fragmentation (Fig. 3B), and a significant reduction of the counts of viable cells (Fig. 3C).

mitoO2−. generated by the short-term coexposure to arsenite and ATP or Cf fails to induce toxicity. (A and B) RP cells exposed for 10 minutes to 2.5 μM arsenite with or without ATP or Cf and analyzed for their caspase 3 activity (A) or for apoptosis, detected by measuring chromatin fragmentation/condensation (B), either immediately or after 6 or 48 hours of growth in fresh culture medium. In other experiments, the cells were grown for 48 hours in the presence of arsenite (A and B). (C) RP cells were treated as indicated above and finally counted after growth in fresh culture medium for increasing time intervals. The closed circles indicate cells grown in the continuous presence of arsenite. (D–F) Cells were loaded (30 minutes) with Fluo 4-AM (D), Rhod-2-AM (E), or DHR (F) and then treated for 10 minutes as indicated in the figure. After treatments, the cells were processed to determine the respective fluorescence responses either immediately or 6 hours postincubation in fresh culture medium. Results represent the means ± S.D. calculated from at least three separate experiments. *P < 0.05; **P < 0.01, as compared with untreated cells. (A, B, and D–F) One-way ANOVA followed by Dunnet’s test; (C) two-way ANOVA followed by Bonferroni’s test.
The short-term exposure paradigm was not associated with an immediate or delayed toxicity as a likely consequence of the reversibility of the upstream effects associated with ROS release. This notion was established by showing that the [Ca2+]c (Fig. 3D), [Ca2+]m (Fig. 3E), and the DHR fluorescence response (Fig. 3F) returned to control levels after 6 hours of post-treatment incubation.
We next performed experiments in which the cells were treated with arsenite/ATP or arsenite/Cf and were then processed for the analysis of DNA strand scission using the alkaline halo assay, a technique developed in our laboratory to detect these lesions at the single-cell level with a sensitivity comparable with that of the comet assay (Guidarelli et al., 2017). Supplemental Figure 4 provides typical images from ethidium bromide–stained cells previously exposed for 30 minutes to arsenite (Supplemental Fig. 4B) or for 10 minutes to ATP (Supplemental Fig. 4C) or Cf (Supplemental Fig. 4D), which all appeared similar to untreated cells (Supplemental Fig. 4A). A typical image of cells with damaged DNA, i.e., displaying evidence for an increased halo/reduced nuclear remnant, was instead obtained from cells exposed for 6 hours to arsenite (Supplemental Fig. 4G). Interestingly, the size of the halo also increased significantly in cells exposed for 30 minutes to the metalloid and ATP (Supplemental Fig. 4E) or Cf (Supplemental Fig. 4F) in the final 10 minutes of incubation.
The time dependence of the DNA strand scission was then measured in cells exposed for increasing time intervals to arsenite, with or without a final 10-minute addition of either ATP or Cf. As shown in Fig. 4A, the nuclear spreading factor slowly increased in cells exposed to the metalloid alone, with kinetics comparable with those describing the rate of ROS formation (Fig. 1A). Accumulation of DNA lesions was instead significantly faster in cells receiving the treatments with the metalloid and ATP or Cf, with a very small delay in comparison with the kinetics of ROS formation detected under the same conditions (Fig. 1A).
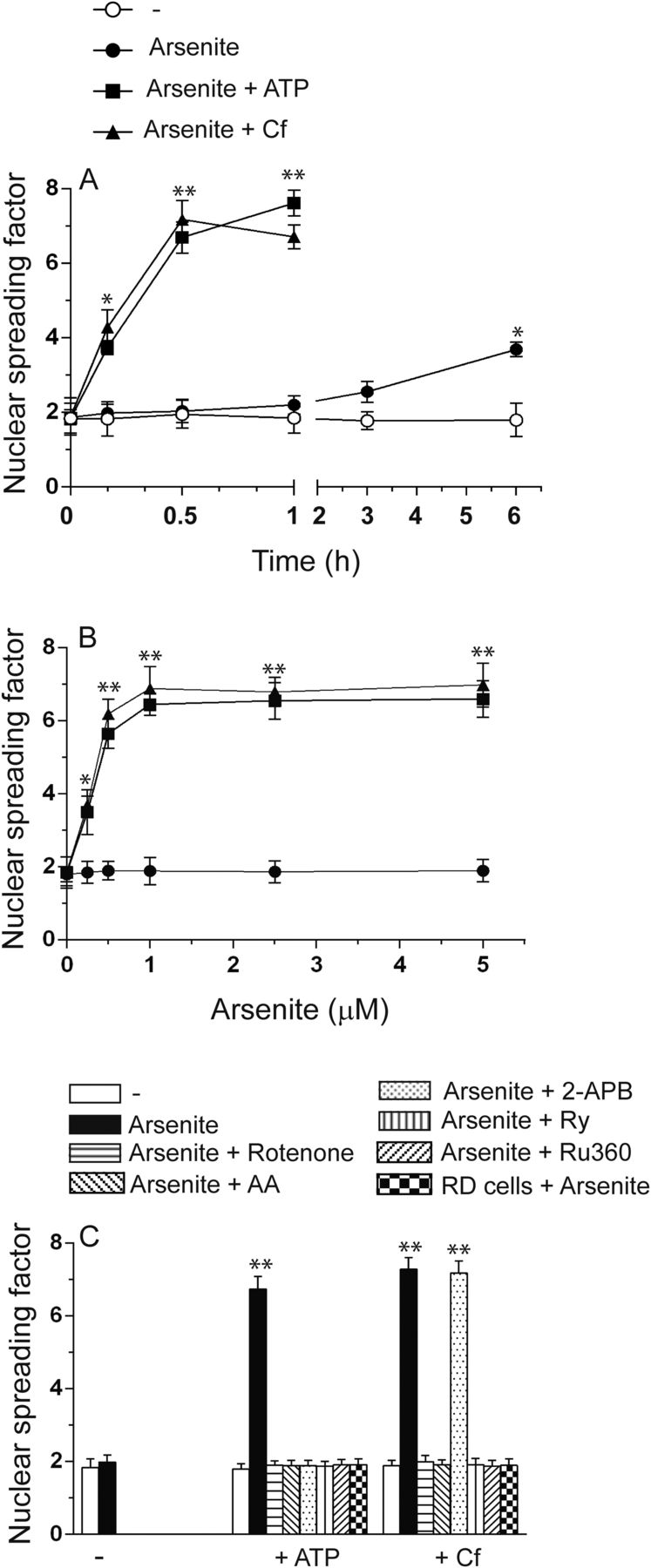
mitoO2−. generated by the short-term coexposure to arsenite and ATP or Cf leads to DNA single-strand breakage. (A) RP cells exposed for increasing time intervals to 2.5 μM arsenite received a 10-minute treatment with ATP or Cf at the end of the incubation and were subsequently analyzed for DNA damage by the alkaline halo assay. (B) Cells exposed for 30 minutes to increasing concentrations of arsenite received ATP or Cf in the last 10 minutes and were then immediately analyzed for DNA damage. (C) Cells were exposed for 5 minutes to the vehicle, rotenone, AA, 2-APB-, Ry, or Ru360 for a further 30 minutes to 2.5 μM arsenite with or without ATP or Cf in the last 10 minutes of incubation and finally analyzed for DNA strand scission. In some experiments, the DNA-damaging response was measured in RD cells coexposed for 30 minutes to arsenite and ATP or Cf. Results represent the means ± S.D. calculated from at least three separate experiments. *P < 0.05; **P < 0.01, as compared with untreated cells. (A and B) Two-way ANOVA followed by Bonferroni’s test; (C) one-way ANOVA followed by Dunnet’s test.
We next established the arsenite concentration dependence for the DNA strand scission observed after 30 minutes with the supplementation of ATP or Cf in the last 10 minutes of incubation. Under these conditions, the metalloid alone failed to produce significant effects (Fig. 4B). Maximal DNA strand scission was instead observed in cells exposed to ≥0.5–1 μM arsenite with ATP or Cf.
Finally, the results illustrated in Fig. 4C indicate that the genotoxic response induced by a 30-minute exposure to 1 µM arsenite associated with ATP or Cf is suppressed by treatments/manipulation abolishing ROS formation at the level of the mitochondrial respiratory chain, i.e., rotenone, AA, and the respiration-deficient phenotype, or preventing mitochondrial Ca2+ accumulation, i.e., Ry and Ru360. 2-APB selectively prevented DNA strand scission in cells exposed to arsenite/ATP.
We therefore conclude that the short-term exposure to arsenite in combination with RyR or IP3R agonists fails to produce immediate or delayed toxicity but nevertheless causes rapid and extensive DNA strand scission entirely attributable to enforced mitochondrial Ca2+ accumulation and Ca2+-dependent mitoO2−. formation.
Discussion
The present study was performed with the aim of investigating the requirements for arsenite in the specific events leading to mitoO2−. formation. For this purpose, we used a mechanism-based approach, taking advantage of a versatile RP cell line characterized by a specific spatial organization of the ER in which arsenite uniquely mediates mitoO2−. formation (Guidarelli et al., 2019a). Based on our recent (Guidarelli et al., 2019a,b) and current findings, this spatial organization of the ER (IP3R vs. RyR) and the close apposition of the RyR with the mitochondria represents a conditio sine qua non for the arsenite-induced mitoO2−. formation. As a consequence, the results obtained using RP cells are likely representative of all cell types responding to arsenite with mitoO2−. formation.
Our previous findings (Fiorani et al., 2018) indicated that exposure of RP cells to 2.5 μM arsenite is associated with a slow and progressive formation of mitoO2−. mediated by at least two separate events. The first one is related to the direct effects of the metalloid on the ER, resulting in Ca2+ mobilization and increased [Ca2+]c (Guidarelli et al., 2018). This response appears slow and requires the micromolar concentration of arsenite apparently needed to target the IP3R and the cross talk between this channel and the RyR (Guidarelli et al., 2018). These responses are then associated with the mitochondrial accumulation of Ca2+ through the MCU, which is necessary but not sufficient for mitoO2−. formation (Guidarelli et al., 2019a). The second event, also necessary but not sufficient for the Ca2+-dependent mitoO2−. formation (Guidarelli et al., 2019a), is instead mediated by arsenite in a mitochondrial target, possibly the respiratory chain, and requires still undetermined durations of exposure to and concentrations of the metalloid.
In the present study, we took advantage of IP3R and RyR agonists to enforce Ca2+ mobilization and then investigated the arsenite requirements for this second effect on mitochondria. Using a general probe, DHR, we obtained evidence for an anticipated ROS response that appeared maximal even when using the 10-minute treatment with 2.5 µM arsenite combined with either ATP or Cf. Furthermore, using the 10-minute coexposure paradigm, we obtained evidence for significant ROS formation, detectable after exposure to 100 nM arsenite and maximal at a 0.5 μM concentration of the metalloid.
These results lead to the conclusion that short exposure times of exposure to low concentrations of the metalloid are required to maximally generate ROS under optimal conditions of Ca2+ availability. As a corollary, the longer times of exposure and higher concentrations of the metalloid normally employed to generate similar effects appear to be uniquely functional to Ca2+ mobilization from the ER.
The second part of this study addressed the mechanism whereby the 10-minute exposure to arsenite and the Ca2+ mobilizing agents mediate the ROS response. In the case of Cf, the cation was derived from the RyR, and mitochondrial Ca2+ accumulation was sensitive to Ry as well as to Ru360. Interestingly, although ATP resulted in a significant increase in the [Ca2+]c through IP3R stimulation (Guidarelli et al., 2019a), it nevertheless caused a mitochondrial accumulation of the cation that was also entirely derived from RyR. Hence, mitochondrial Ca2+ accumulation was also sensitive to Ry and Ru360, as in the case of Cf.
These results indicate that the effects of the IP3R and RyR agonists are dependent on their ability to induce mitochondrial Ca2+ accumulation. The fact that in these cells the RyR is localized in close apposition with the mitochondria (Guidarelli et al., 2019a) favors extensive mitochondrial Ca2+ accumulation under conditions in which the cation is released by the RyR (Eisner et al., 2013; Fauconnier et al., 2013; Delmotte and Sieck, 2015; Raffaello et al., 2016; Hirabayashi et al., 2017).
These observations also provided the experimental ground to determine that ROS formation was mediated by the increased [Ca2+]m, with hardly any role detected for the increased [Ca2+]c. Indeed, the DHR fluorescence response mediated by arsenite/Cf or ATP was sensitive to treatments preventing mitochondrial Ca2+ accumulation and independent on the [Ca2+]c. For example, ATP produced a greater increase in [Ca2+]c than Cf, but their effects on ROS formation were identical. Most importantly, Ru360 in both circumstances prevented the increase in [Ca2+]m and ROS formation but did not produce effects on the [Ca2+]c.
The notion that ROS formation is mediated by the increased [Ca2+]m may indicate that ROS are produced within these organelles. This notion was established by showing that the DHR fluorescence response evoked by arsenite/Cf or arsenite/ATP was sensitive to rotenone, the respiration-deficient phenotype, or AA. Note that RP cells express high-affinity sodium-dependent vitamin C transporter 2(Km = 26.96 ± 1.46 µM) in their plasma and mitochondrial membranes (Fiorani et al., 2015). It follows that in these cells, exposure to suboptimal concentrations of AA, lower than/close to the Km for sodium-dependent vitamin C transporter 2 leads to the accumulation of low cytosolic and very high mitochondrial levels of the vitamin, thereby providing an excellent tool to selectively scavenge mitoO2−. (Cantoni et al., 2018).
The formation of mitoO2−. in cells exposed to arsenite/ATP or arsenite/Cf was also demonstrated in experiments in which MitoSOX red, which only detects mitochondrial ROS (Mukhopadhyay et al., 2007), was used in the place of DHR. Using this probe, we could recapitulate all the results obtained with DHR, including the inhibitory effects mediated by agents resulting in mitochondrial Ca2+ accumulation, as well as by treatments/conditions impairing the ability of mitochondria to generate ROS.
The above findings therefore convincingly demonstrate that mitoO2−. is the only species generated by arsenite/ATP or arsenite/Cf, a conclusion ruling out the possible involvement of NADPH oxidase(s). However, since this system is considered of pivotal importance in arsenite toxicology (Chou et al., 2004; Straub et al., 2008; Guidarelli et al., 2016b) and since ATP promotes a large increase in [Ca2+]c, we nevertheless considered it important to perform experiments directly addressing this possibility. In this direction, it is informative to remember that RP cells express the cytosolic isoform (p47phox and p67phox) of NADPH oxidase, which besides being responsive to PMA-dependent stimulation also responds to high arsenite concentrations (Guidarelli et al., 2016b, 2019b; Fiorani et al., 2018). We found that NADPH oxidase inhibitors, while suppressing the PMA-induced DHR fluorescence response, produced hardly any effect on the DHR fluorescence response induced by arsenite/ATP. In addition, PMA, unlike arsenite/ATP, promoted significant p47phox phosphorylation, thereby ruling out the possibility of a contribution of NADPH oxidase in the overall ROS response induced by arsenite/ATP.
In the third part of this study, we determined that combined short-term exposure to arsenite and ATP or Cf fails to produce effects on the intracellular levels of GSH or ATP. Likewise, there was no evidence of detectable Trx2 oxidation, thereby implying that mitochondrial O2−. and H2O2 are effectively scavenged by the mitochondrial antioxidant network. Finally, we also failed to detect delayed toxic effects eventually leading to mitochondrial dysfunction and apoptosis.
The absence of the above responses/consequences is most likely attributable to the transient nature of the effects mediated by the metalloid in combination with the Ca2+ mobilizing agent. Consistently, post-treatment incubation in fresh culture medium allowed complete recovery of the [Ca2+]c and [Ca2+]m and returned the rate of mitoO2−. formation to control levels.
The same requirements of a prolonged exposure to arsenite alone to promote toxicity were recently established in our laboratory (Guidarelli et al., 2015, 2016a). Indeed, a 6-hour exposure to arsenite followed by post-treatment incubation in fresh culture medium failed to bear deleterious consequences, which instead occurred in cells that were continuously exposed to the metalloid and experienced a sustained increase in [Ca2+]c and [Ca2+]m and persistent mitoO2−. formation, mitochondrial dysfunction, and apoptosis (Guidarelli et al., 2019a).
It therefore appears that the process of transient mitoO2−. formation encompassed by the low arsenite concentration under conditions of enforced mitochondrial Ca2+ accumulation is devoid of intrinsic cytotoxicity. Hence, it was interesting to observe that these same conditions were associated with the occurrence of an event relevant for arsenic-induced carcinogenesis. We found that the Ca2+ mobilizing agents caused a remarkable anticipation of the DNA-damaging response evoked by arsenite. These kinetics, although slightly slower, were nevertheless comparable with those observed in experiments measuring ROS formation, which is clearly expected since a cause-effect relationship links these parameters. Consistently, a remarkable leftward shift was observed in the dose-response curves for arsenite-induced DNA strand scission after a very short time of exposure.
In conclusion, the results presented in this study provide solid experimental evidence that short-term exposure to low concentrations of arsenite is required to promote the mitochondrial events associated with Ca2+-dependent mitoO2−. formation. This is in contrast with the more stringent requirements identified for the upstream Ca2+ mobilization, likely representing a critical limiting factor involved in the regulation of the arsenite-dependent ROS formation; that is, the presence of the metalloid is obviously necessary, but in fact the concentration and time requirements are very low when Ca2+ is present. A scheme summarizing these findings is shown in Fig. 5.

Arsenite requirements for maximal Ca2+-dependent mitoO2−./H2O2 formation. Prolonged (hours) exposure to high concentrations of arsenite is necessary to target the IP3R and the cross talk between this pool and the RyR to promote RyR-derived Ca2+ accumulation in mitochondria. These events are favored by the specific architecture of the ER and by the close apposition between the RyR and mitochondria of RP cells. Remarkably shorter exposure to significantly lower concentrations of the metalloid are instead required to maximally generate mitoO2−. under optimal conditions of Ca2+ availability. This notion was clearly established by enforcing mitochondrial Ca2+ accumulation with IP3R or RyR agonists, and the resulting mitoO2−. and its dismutation product, H2O2, although failing to promote mitochondrial dysfunction and MPT-dependent apoptosis, nevertheless caused extensive DNA strand scission. MPT, mitochondrial permeability transition.
Understanding the specific requirements of arsenite to induce its effects on Ca2+ homeostasis is of particular importance to determine the threshold limits of exposure to this dangerous environmental contaminant. It is indeed important to consider the possibility that the effects of arsenite are concomitant with those of other substances, e.g., pharmaceuticals, environmental contaminants, and food additives, as well as hormones and mediators, including those released under inflammatory conditions, which may enforce the mitochondrial accumulation of Ca2+, thereby fostering the effects of arsenite on mitoO2−. formation.
The present study, while providing important details on the mechanism whereby arsenite promotes mitoO2−. formation, raises concerns on possible effects mediated by apparently low levels of the metalloid and the concomitant activation of physiologic, pathologic, and toxicologic responses leading to enforced mitochondrial Ca2+ accumulation.
Authorship Contributions
Participated in research design: Guidarelli, Cerioni, Fiorani, Catalani, Cantoni.
Conducted experiments: Guidarelli, Cerioni, Fiorani, Catalani.
Performed data analysis: Guidarelli, Cerioni, Fiorani, Catalani, Cantoni.
Wrote or contributed to the writing of the manuscript: Guidarelli, Fiorani, Cantoni.
Footnotes
- Received September 16, 2019.
- Accepted January 13, 2020.
This work was supported by Ministero dell’Università e della Ricerca Scientifica e Tecnologica, Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale, 2017, [Grant number: 2017FJSM9S] and by Dipartimento di Scienze Biomolecolare, Università degli Studi di Urbino Carlo Bo, Progetti Valorizzazione (2017–2018).
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- 2-APB
- 2-aminoethoxydiphenyl borate
- AA
- L-ascorbic acid
- AM
- acetoxymethyl ester
- Cf
- caffeine
- [Ca2+]c
- cytosolic Ca2+ concentration
- [Ca2+]m
- mitochondrial Ca2+ concentration
- DHR
- dihydrorhodamine 123
- DPI
- diphenyleneiodonium
- ER
- endoplasmic reticulum
- GSH
- glutathione
- HPLC
- high-performance liquid chromatography
- IP3R
- inositol-1,4,5-trisphosphate receptor
- MCU
- mitochondrial Ca2+ uniporter
- mitoO2−.
- mitochondrial O2−.
- O2−.
- superoxide
- PMA
- phorbol 12-myristate 13-acetate
- p47phox
- p47 phagocyte oxidase
- RD cell
- respiration-deficient cell
- ROS
- reactive oxygen species
- RP cell
- respiration-proficient cell
- Ry
- ryanodine
- RyR
- Ry receptor
- Trx2
- thioredoxin-2
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}