


Abstract
The imidizodiazepine, 5-(8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)oxazole (KRM-II-81), is selective for α2/3-containing GABAA receptors. KRM-II-81 dampens seizure activity in rodent models with enhanced efficacy and reduced motor-impairment compared with diazepam. In the present study, KRM-II-81 was studied in assays designed to detect antiepileptics with improved chances of impacting pharmaco-resistant epilepsies. The potential for reducing neural hyperactivity weeks after traumatic brain injury was also studied. KRM-II-81 suppressed convulsions in corneal-kindled mice. Mice with kainate-induced mesial temporal lobe seizures exhibited spontaneous recurrent hippocampal paroxysmal discharges that were significantly reduced by KRM-II-81 (15 mg/kg, orally). KRM-II-81 also decreased convulsions in rats undergoing amygdala kindling in the presence of lamotrigine (lamotrigine-insensitive model) (ED50 = 19 mg/kg, i.p.). KRM-II-81 reduced focal and generalized seizures in a kainate-induced chronic epilepsy model in rats (20 mg/kg, i.p., three times per day). In mice with damage to the left cerebral cortex by controlled-cortical impact, enduring neuronal hyperactivity was dampened by KRM-II-81 (10 mg/kg, i.p.) as observed through in vivo two-photon imaging of layer II/III pyramidal neurons in GCaMP6-expressing transgenic mice. No notable side effects emerged up to doses of 300 mg/kg KRM-II-81. Molecular modeling studies were conducted: docking in the binding site of the α1β3γ2L GABAA receptor showed that replacing the C8 chlorine atom of alprazolam with the acetylene of KRM-II-81 led to loss of the key interaction with α1His102, providing a structural rationale for its low affinity for α1-containing GABAA receptors compared with benzodiazepines such as alprazolam. Overall, these findings predict that KRM-II-81 has improved therapeutic potential for epilepsy and post-traumatic epilepsy.
SIGNIFICANCE STATEMENT We describe the effects of a relatively new orally bioavailable small molecule in rodent models of pharmaco-resistant epilepsy and traumatic brain injury. KRM-II-81 is more potent and generally more efficacious than standard-of-care antiepileptics. In silico docking experiments begin to describe the structural basis for the relative lack of motor impairment induced by KRM-II-81. KRM-II-81 has unique structural and anticonvulsant effects, predicting its potential as an improved antiepileptic drug and novel therapy for post-traumatic epilepsy.
Introduction
Epilepsy is a chronic and highly prevalent neurologic disorder that affects millions of people world wide (Sadr et al., 2018). The disease has serious consequences for the life of the affected individual, which includes lethality (DeGiorgio et al., 2019). Although there are many anticonvulsant drugs approved to decrease seizure probability, seizures are not fully controlled in many patients despite polypharmacy approaches. In 60%–70% of patients (Marson et al., 2007) existing drugs are not efficacious at some point in the disease progression (Sinha and Siddiqui, 2011; Banerjee et al., 2014), and the lack of seizure protection places the patient at increased risk. As such, pharmaco-resistant epilepsy patients sometimes require invasive therapeutic options such as surgical resection or disconnection (Hwang and Kim, 2019). Therefore, the need for improved antiepileptic drugs is mandated and continues to generate continuous vigilance by researchers (Barker-Haliski et al., 2017) and patient advocacy communities (e.g., http://advocacy.epilepsy.com).
Although enhancing GABAergic inhibitory tone is a well-established mechanism for seizure protection with multiple anticonvulsant drugs approved in this class (White et al., 2007; Gitto et al., 2010), recent attention has been directed toward GABA modulators that have reduced side-effect liabilities. In particular, the potential advantages of developing positive allosteric modulators (PAMs) of α2/3-containing GABAA receptors as antiepileptic drugs has come to the forefront in the last 2 years. The potential advantages of this mechanism over that of nonselective PAMs (e.g., diazepam) include lower risk for somnolence, motor impairment, and abuse compared with nonselective GABAA receptor PAMs (Ator et al., 2010; Atack, 2011; Ralvenius et al., 2015; Zuiker et al., 2016; Witkin et al., 2018; Moerke et al., 2019). Reduced motor impairment enables the possibility of enhancing anticonvulsant efficacy by relieving the patients of this dose-limiting side effect.
5-(8-Ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)oxazole (KRM-II-81) is an orally bioavailable compound with selectivity for α2/3-containing GABAA receptors. KRM-II-81 reduces seizures in rodent models induced by acute and chronic seizure provocation (Witkin et al., 2018). KRM-II-81 was often more efficacious and produced less motor impairment than the non-α2/3-selective anticonvulsant, diazepam (Witkin et al., 2018). The present series of experiments was undertaken to increase the understanding of the range of conditions under which KRM-II-81 will function as an anticonvulsant. Animal models are known to have predictive validity for detecting drugs that treat epilepsies (Simonato et al., 2014). In addition to primary screening models, other more complex animal models have been developed to further refine predictive efficacy and differentiation (Barker-Haliski et al., 2017).
One specific aim of the current research was to evaluate the potential efficacy of KRM-II-81in pharmaco-resistant epileptic patients. It had already been shown that KRM-II-81 was active in the 6 Hz mouse model (Witkin et al., 2018) which is used to predict novel treatments (Barton et al., 2001) and potential agents for pharmaco-resistant epilepsies (Wilcox et al., 2013; Leclercq et al., 2014). A rat model using 6 Hz stimulation is also in development (Metcalf et al., 2017). Several additional rodent seizure models used in antiepileptic drug screens to help guide new molecules into the pharmaco-resistant epilepsy therapy space (Wilcox et al., 2013; Barker-Haliski et al., 2017) were studied here: the mesial temporal lobe epilepsy model in mice, lamotrigine-resistant kindling model in rats, and kainate-induced chronic epilepsy model in rats. The mesial temporal lobe epilepsy model is a model of partial seizures initially triggered by kainate injections into the dorsal hippocampus. The mice then develop enduring epileptic events that are characteristic of temporal lobe epilepsy in patients (Bouilleret et al., 1999; Riban et al., 2002). In the lamotrigine-resistant kindling model, kindling current is delivered in the presence of lamotrigine, rendering the rats insensitive to the anticonvulsant effects of lamotrigine and other drugs (Srivastava et al., 2013; Wilcox et al., 2013). The model is thus used in advanced antiepileptic drug screening to identify new drugs for pharmaco-resistant epilepsy (Wilcox et al., 2013; Barker-Haliski et al., 2017). In the kainate-induced chronic epilepsy model, repeated low doses of kainate initiate long-term sequalae of focal and generalized seizures. This model enables the monitoring of spontaneous recurrent seizures and clinically relevant measures of epilepsy. It is used in late stages of antiepileptic drug screening (Barker-Haliski et al., 2017; Grabenstatter and Dudek, 2019) to create clinically relevant measures of epilepsies for antiepileptic drug differentiation (West et al., 2018).
Another specific aim of the present study was to assess whether KRM-II-81 might have a role in mitigating seizure-related phenomenon in traumatic brain injury (TBI). Seizure probabilities and the development of epilepsy (post-traumatic epilepsy) are increased in cases of TBI. Early after trauma, the probability of seizures is relatively high, making anticonvulsant treatment a relatively standard procedure despite only small proven efficacy (Wat et al., 2019). Although seizure likelihood decreases after the first few weeks of injury, post-traumatic epilepsy develops later in TBI patients and is associated with affective, neurocognitive, and psychosocial disruption of life (Semple et al., 2019). Ping and Jin (2016) described the sequalae of events post TBI using a mouse model. They reported an early quiescence phase of neuronal activity, followed by sustained hyperactivity of cortical neurons after controlled cortical impact (CCI) injury that they attributed to homeostatic compensation. These mice with TBI are at increased risk in developing post-traumatic epilepsy (Bolkvadze and Pitkänen, 2012). We hypothesized that the late-phase cortical hyperactivity, a marker of injured brain at risk for seizures, would be suppressed by KRM-II-81. We conducted such an experiment in the present study with the idea of further defining methods to decrease seizure risk post TBI for which few treatment options exist (Temkin, 2009).
Materials and Methods
Compounds
KRM-II-81 was synthesized by us (JM Cook Laboratory) as previously described (Poe et al., 2016; Li et al., 2018). The other compounds were obtained from Sigma-Aldrich (St. Louis, MO). KRM-II-81was suspended in 1% carboxymethylcellulose and dosed at 1 ml/kg in rats below doses of 30 mg/kg, at 3 ml/kg for doses of 30 mg/kg, and at 6 ml/kg for doses of 60 mg/kg. Mice were given 10 ml/kg.
Rodent Assays
All studies were performed in accordance with guidelines of the National Institutes of Health and by local animal care and use committees. The local animal care and use committee and veterinary staff provided direct oversight of the animals by inspections, protocol reviews, laboratory site visits, and animal health monitoring.
Corneal Kindling
In the corneal kindling experiments, KRM-II-81 was evaluated for its ability to block seizures in fully kindled mice. Adult male CF1 mice (n = 8 per group, 18–25 g) were kindled using methods previously described (Matagne and Klitgaard, 1998; Rowley and White, 2010). They were kindled to a criterion of five consecutive secondarily generalized seizures of stage 5 (Racine, 1972). Briefly, mice were exposed to twice daily corneal stimulation (3 mA at 60 Hz for 3 seconds). After reaching the first stage 5 seizure (generally after about 2 weeks), the twice daily stimulation regimen was continued until each mouse achieved the criteria of five consecutive stage 5 seizures, at which point the mouse was considered to be fully seizure kindled. Fully kindled mice continued to be stimulated every 2 to 3 days until all mice became fully kindled.
Testing of KRM-II-81 began at least 5 days after the last corneal stimulation. KRM-II-81 was dosed orally 2 hours prior to stimulation for the assessment of anticonvulsant efficacy. Doses of 1–30 mg/kg were tested in groups of eight mice. Doses were selected based on the efficacy of KRM-II-81 studied in other seizure models by this route, where 10 mg/kg was a minimal effective dose (Witkin et al., 2018). Oral dosing was used here to define oral efficacy based on the oral dosing exposure of KRM-II-81 (Poe et al., 2016; Witkin et al., 2019ba). The percentage of mice protected and the seizure severity score (Racine, 1972) were measured.
The effects of KRM-II-81 on seizure protection were assessed by Fisher’s exact probability test and the ED50 and 95% confidence limits were determined by probit analysis. Effects of KRM-II-81 on seizure severity scores were analyzed by one-way ANOVA followed by Dunnett’s test. Probabilities of < 0.05 were set a priori as the accepted level of statistical significance.
Mesial Temporal Lobe Model
Seizures were generated by unilateral application of kainic acid into the dorsal hippocampus as previously described (Bouilleret et al., 1999; Riban et al., 2002) to induce seizure discharges that are characteristics of human temporal lobe epilepsy. Then, 2 to 3 weeks after the kainate injection, spontaneous recurrent hippocampal paroxysmal discharges were recorded in the epileptic hippocampus, where they remain stable and for the life of the mouse. Briefly, adult male C57/Bl6 mice were stereotaxically injected (100 nl) with kainate (1 nmol) and implanted with a single bipolar electrode in the dorsal hippocampus. The mice were allowed to recover for 4 weeks prior to drug testing. Each mouse was used as its own control. Digital electroencephalogram (EEG) recordings were performed on freely moving animals for 20 minutes prior to drug testing and then for 90 minutes postdosing.
KRM-II-81 was studied at 15 mg/kg by the oral route dosed 2 hours prior to the seizure sampling period. The dose was chosen based on minimal effective doses observed in other rodent seizure models (Witkin et al., 2018). The oral route was used to determine oral activity of KRM-II-81 as in the corneal kindling model described previously. The number of hippocampal paroxysmal discharges was counted. The effects of KRM-II-81 were studied in four mice. The number of hippocampal paroxysmal discharges at baseline and after 2 hours were compared by a two-tailed, Student’s t test for paired observations. Probabilities < 0.05 were set a priori as the accepted level of statistical significance.
Lamotrigine-Resistant Kindling Model
Rats were seizure kindled in the presence of lamotrigine to create seizures that are not responsive to blockade by lamotrigine and some other anticonvulsants (Srivastava et al., 2013). The rats were prepared as previously described (Srivastava et al., 2013; Wilcox et al., 2013). Briefly, anesthetized male Sprague-Dawley rats weighing about 250–300 g were surgically implanted with an electrode into the left amygdala (anterio-posterior +5.7 mm, medio-lateral +4.5 mm, and dorso-ventral +2.0 mm from intra-aural zero). Then, the rats were returned to their home cages for 1 week before starting the kindling process. Kindling was achieved through daily stimulations. The kindling procedure consisted of delivery of a 200 μA stimulus (suprathreshold) daily until all animals in both treatment groups displayed consistent stage 4 or 5 seizures. One week after all rats were kindled, rats were given a challenge dose of lamotrigine (30 mg/kg, i.p.) before being stimulated to confirm the ability of lamotrigine to block seizures in the vehicle-kindled rats and the lack of blockade in the lamotrigine-kindled rats. A 3-day rest period was then implemented, and on day 3 the rats were stimulated to verify that they were fully seizure kindled.
KRM-II-81 testing began the next day. KRM-II-81 was given by intraperitoneal injection 60 minutes prior to electrical stimulation. Tests with KRM-II-81 were scheduled only after at least three drug-free days. KRM-II-81 was studied in doses of 0 (vehicle), 1, 5, 10, 20, and 40 mg/kg in eight rats per dose. Doses were selected based on the efficacy of KRM-II-81 studied in other seizure models by this route, where 10 mg/kg was a minimal effective dose (Witkin et al., 2018). The following measures of seizure activity were recorded: the number of rats not convulsing poststimulation (Racine score < 3), the seizure severity score, and the seizure duration. The effects of KRM-II-81 on seizure protection were assessed by Fisher’s exact probability test and the ED50 and 95% confidence limits were determined by probit analysis. The effects of KRM-II-81 on seizure severity scores were analyzed by one-way ANOVA followed by Dunnett’s test. Probabilities < 0.05 were set a priori as the accepted level of statistical significance.
Kainate-Initiated Chronic Epilepsy
Induction of a state of chronic epilepsy in rats was produced by repeated low doses of kainic acid. The procedure results in the occurrence, weeks later, of recurrent spontaneous focal and generalized seizures . The methods are as described previously (Barker-Haliski et al., 2017; Grabenstatter and Dudek, 2019). Briefly, male Sprague-Dawley rats were injected with 7.5 mg/kg kainic acid, i.p., at 0 hour, 1 hour, and every subsequent half-hour (up to 4 hours), or until the animal displayed two Racine stage 5 seizures and was then given 3 ml of Ringers solution to prevent dehydration.
Ten weeks after kainic acid, the rats are implanted with Millar wireless telemeters implanted into the peritoneal space. The EEG cable was routed from the stomach to the head underneath the skin. Three holes were drilled in the skull and three fixation screws were placed. Two additional holes were drilled, and an EEG wire was placed in each and secured by super glue. Rats were placed in a living environment for EEG monitoring. Rats with the highest spontaneous seizure rates were used for drug testing.
Chronic seizure monitoring was performed in 12 rats over 21 days. KRM-II-81 was studied at 20 mg/kg, i.p., three times per day, based on kinetic estimates of drug exposure in plasma and brain (Poe et al., 2016) and the efficacy of this dose range in other seizure models (Witkin et al., 2018). The intraperitoneal route was used for ease of dosing over this time period. In the first week of seizure monitoring, a baseline seizure rate was determined. During the following days, injections were given over 5 days (Monday to Friday), where a crossover design was implemented for vehicle or KRM-II-81 such that all rats received baseline, vehicle, and drug treatments.
EEG data were reviewed daily by a reviewer blinded to treatment. The reviewer scored events from automatically generated lists of potential seizure events collected each night by an automated seizure detection algorithm. Both the percentage of mice that were seizure free and the seizure burden scores were calculated. Seizure burden scores were calculated as the sum total of all seizure scores divided by the number of days tested. The seizure-free scores (number of rats not exhibiting seizure events/12 rats tested) were analyzed by Fisher’s exact probability test. The seizure burden scores were analyzed by Wilcoxon rank sum test. Probabilities < 0.05 were set a priori as the accepted level of statistical significance.
Traumatic Brain Injury
Preparations of Cranial Window and Controlled Cortical Impact Model.
Adult GCaMP6s transgenic mice (2.5–3 months old) were anesthetized with ketamine/xylazine (87.7/12.3 mg/kg, i.p.). Large cranial windows were prepared using a previously described technique (Kim et al., 2016). Briefly, the mice were fixed on a stereotaxic apparatus. Following exposure of the skull with a midline incision and removal of skin, an area of skull (7.5 mm long and 8 mm wide) covering both cortical hemispheres was drilled and carefully removed. After the cortex stopped bleeding, a CCI injury was made using a previously published technique (Ping et al., 2014). The CCI was induced on the exposed left sensorimotor cortex by using a 2-mm-diameter impacting rod tip to compress the cortex at a velocity of 3.0 m/s to a depth of 0.5 mm. The cranial window was then covered with a piece of curved cover glass and the edge was sealed by applying super glue. The mice in the sham group received only craniotomy without CCI.
In Vivo Two-Photon Imaging for Evaluating Neuronal Activity.
The mice were sedated with chlorprothixene and calcium transients of cortical layer II/III neurons were imaged at two to four frames per second for 2 minutes using a two-photon microscope (Prairie Technologies) (Xiong et al., 2017). A tunable Maitai titanium:sapphire laser (Newport, Mountain View, CA) was tuned to 900 nm to provide excitation. Band-pass–filtered emitting fluorescence (560–660 nm) was collected by photomultiplier tubes in the system. The same imaging field was repeatedly imaged during an imaging session. KRM-II-81 was given intraperitoneally at 10 mg/kg based on the minimal effective dose reported in other seizure models (Witkin et al., 2018). Imaging of cortical activity was done for 4 hours, thus obtaining data for 0.5, 1, 2, and 4 hours postdosing.
For data analysis, neurons were manually selected based on average projections of image stacks combined with quickly examining the whole image stacks. Raw fluorescence (F) was measured as the average fluorescence of pixels lying within a cell body in each frame and background fluorescence (Fo) was measured as the mean of two sites that were just around the cell and contained no neuronal structures. The ΔF/F signals were calculated as (F − Fo)/Fo. Cell activity was measured by both the mean integrated fluorescence and ratio of active neurons (Xiong et al., 2017). Integrated fluorescence was calculated by integrating all fluorescence above a threshold of 20% ΔF/F. The threshold was used to eliminate the possible influence of noise. Active neurons were defined as cells that had fluorescence intensity above the threshold level at least once. The ratio of active neurons at a given time period was calculated by dividing the number of active cells by the total number of cells in that imaging field (Xiong et al., 2017). Both values of each mouse at different time points were normalized to the baseline levels. Data were analyzed by one-way ANOVA followed by Bonferroni test. Probabilities < 0.05 were set a priori as the accepted level of statistical significance.
Molecular Structural Modeling
Ligand-protein interactions were analyzed by molecular docking using AutoDock Vina 1.5.6 (Trott and Olson, 2010). The Protein Data Bank (PDB) file of the CryoEM structure of the human full-length α1β3γ2L GABAA receptor in complex with diazepam (PDB: 6HUP) and alprazolam (PDB: 6HUO) (Masiulis et al., 2019) was downloaded from the Protein Data Bank and prepared for docking by fixing missing bonds or atoms, adding polar hydrogens and assigning charges by AM1-BCC (Austin Model1 with bond charge correction), and removing water molecules. The proteins were validated by first removing the bound ligand (diazepam or alprazolam), followed by docking it in the same binding site. The compounds were drawn and energy minimized in Chimera (Duncalfe et al., 1996). A grid size of 15 × 15 × 15 Å of the 6HUP PDB structure was used, centered at coordinates 117.23 (x), 157.48 (y), and 110.75 (z). For the 6HUO PDB structure, a grid size of 15.78 × 16.41 × 14.34 Å was used, centered at coordinates 152.80 (x), 163.02 (y), and 161.14 (z). Illustrations of the three-dimensional models were generated using Chimera (Pettersen et al., 2004) and Python (Sanner, 1999). Dockings were performed with standard search parameters and poses with the best scores were selected for the analysis.
Side-Effect Evaluations
Since the molecular model can be used to describe the mechanistic rationale for the relatively mild motor-impacting effects (e.g., sedation and ataxia) of KRM-II-81 compared with diazepam or alprazolam as previously described (Witkin et al., 2018), we studied the effects of a very high dose of KRM-II-81. Male Sprague-Dawley rats (n = 8) orally given 300 mg/kg KRM-II-81 were observed for signs of behavioral toxicity (sedation, ataxia, righting response, etc.) at 0.25, 0.5, 1, 2, 4, and 6 hours postdosing.
Results
Corneal Kindling
Orally administered KRM-II-81 (2 hours prior) increased the percentage of seizure-kindled mice that were protected from corneal stimulation (Fig. 1, inset). The minimal effective dose of KRM-II-81 was 8 mg/kg with full protection produced by doses of 15, 25, and 30 mg/kg. The ED50 value for protection was 3.96 mg/kg (95% confidence limits: 2.11–6.05). The severity of the seizures produced in corneal-kindled mice was also reduced by KRM-II-81 with a minimal effective dose and a fully protective dose identical to that observed with the measure of seizure prevalence (F5,47 = 24.0, P < 0.0001) (Fig. 1, left panel).
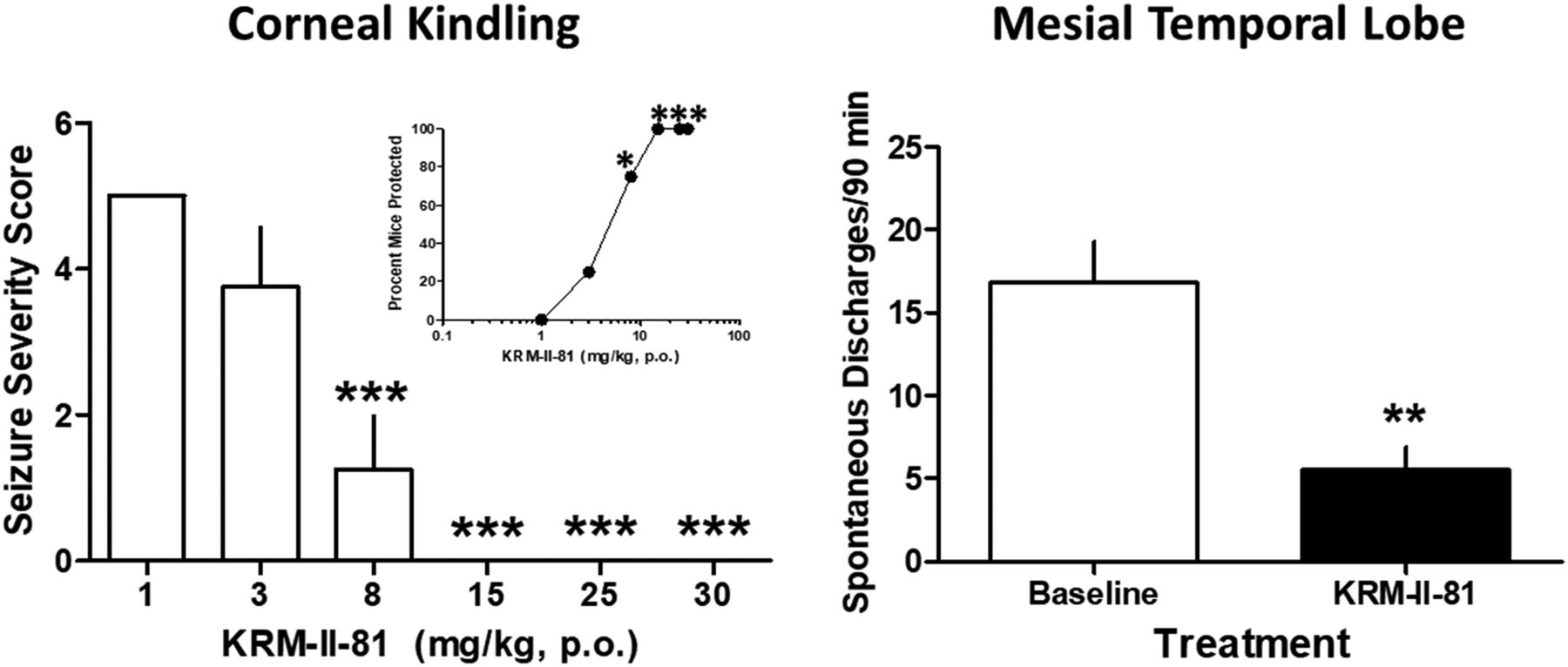
Effects of KRM-II-81 in corneal-kindled mice (left) and the mesial temporal lobe epilepsy model in mice (right). (Left panel) KRM-II-81 was given orally, 2 hours prior to testing in corneal-kindled mice. KRM-II-81 dose dependently increased the percentage of mice that were protected against convulsions (inset) and decreased seizure severity in the mice. Each point or bar represents the mean ± S.E.M. of eight mice. Seizure prevalence (inset) was assessed with Fisher’s exact probability test with *P < 0.05 with vehicle-treated mice. Seizure severity was evaluated with ANOVA followed by Dunnett’s post-hoc test with ***P < 0.001 compared with vehicle control mice. (Right panel) Mice with kainate-induced mesial temporal lobe seizures exhibited spontaneous recurrent hippocampal paroxysmal discharges. KRM-II-81 (15 mg/kg, orally, 2 hours prior) was tested in four mice (t3 = 8.6, P < 0.01 as indicated by **).
Mesial Temporal Lobe Model
Mice with kainate-induced mesial temporal lobe seizures exhibited spontaneous recurrent hippocampal paroxysmal discharges. These discharges occurred from 30 to 60 times per hour when the animals were in a state of quiet wakefulness. They lasted about 15–20 seconds, where they were associated with behavioral arrest and/or mild motor automatisms. The baseline levels of spontaneous discharges were 16.8 ± 2.5. In the presence of 15 mg/kg KRM-II-81 (given orally, 2 hours prior), there was significant reduction in spontaneous discharges to a mean value of 5.5 ± 1.4 (t3 = 8.6, P < 0.01) (Fig. 1, right panel).
Lamotrigine-Resistant Kindling Model
Electrical kindling of the basolateral amygdala in the presence of lamotrigine induced full-blown stage 5 seizures in rats. KRM-II-81 was given in doses of 1–40 mg/kg, i.p. (1 hour prior to stimulation), in fully kindled rats. KRM-II-81 produced a dose-dependent increase in the number of rats protected from seizures (Table 1) with a minimal effective dose of 20 mg/kg. The ED50 value for protection was 19.2 (95% confidence limits: 9.6–56) mg/kg. KRM-II-81 also produced dose-dependent protection against the severity (Table 1) and duration (Table 1) of seizures in these rats with minimal effective doses of 5 and 10 mg/kg for these measures, respectively. One-way ANOVA confirmed significant protection conferred by KRM-II-81 against both seizure severity (F5,47 = 18.1, P < 0.0001) and seizure duration (F5,47 = 45.9, P < 0.0001).
Effects of KRM-II-81 on seizures induced by electrical stimulation of the basolateral amygdala in rats that were seizure kindled in the presence of lamotrigine
KRM-II-81 was given intraperitoneally (1 hour prior to testing) in eight rats. Seizure prevalence was assessed with Fisher’s exact probability test with *P < 0.05 and ** P <0.01 compared with vehicle-treated rats. Seizure severity and duration were evaluated with ANOVA followed by Dunnett’s post-hoc test with * P < 0.05 and ***P < 0.001 compared with vehicle control rats.
Kainate-Induced Chronic Epilepsy
Rats previously given repeated low-dose exposures to kainate developed stage 5 seizures and were then tested under baseline, drug, or vehicle dosing conditions 10 weeks post kainate-induced chronic epilepsy. The 12 rats studied showed high levels of baseline seizures (focal and generalized) over the 21-day observation period (Fig. 2; Table 2). When KRM-II-81 was dosed at 20 mg/kg, i.p. (three times per day), rats exhibited decreases in seizure burden by 4-fold; seizures re-emerged when drug was removed (Fig. 2; Table 2). KRM-II-81 also produced increases from 8% to 50% in the number of rats that were seizure free (Fig 2; Table 2).
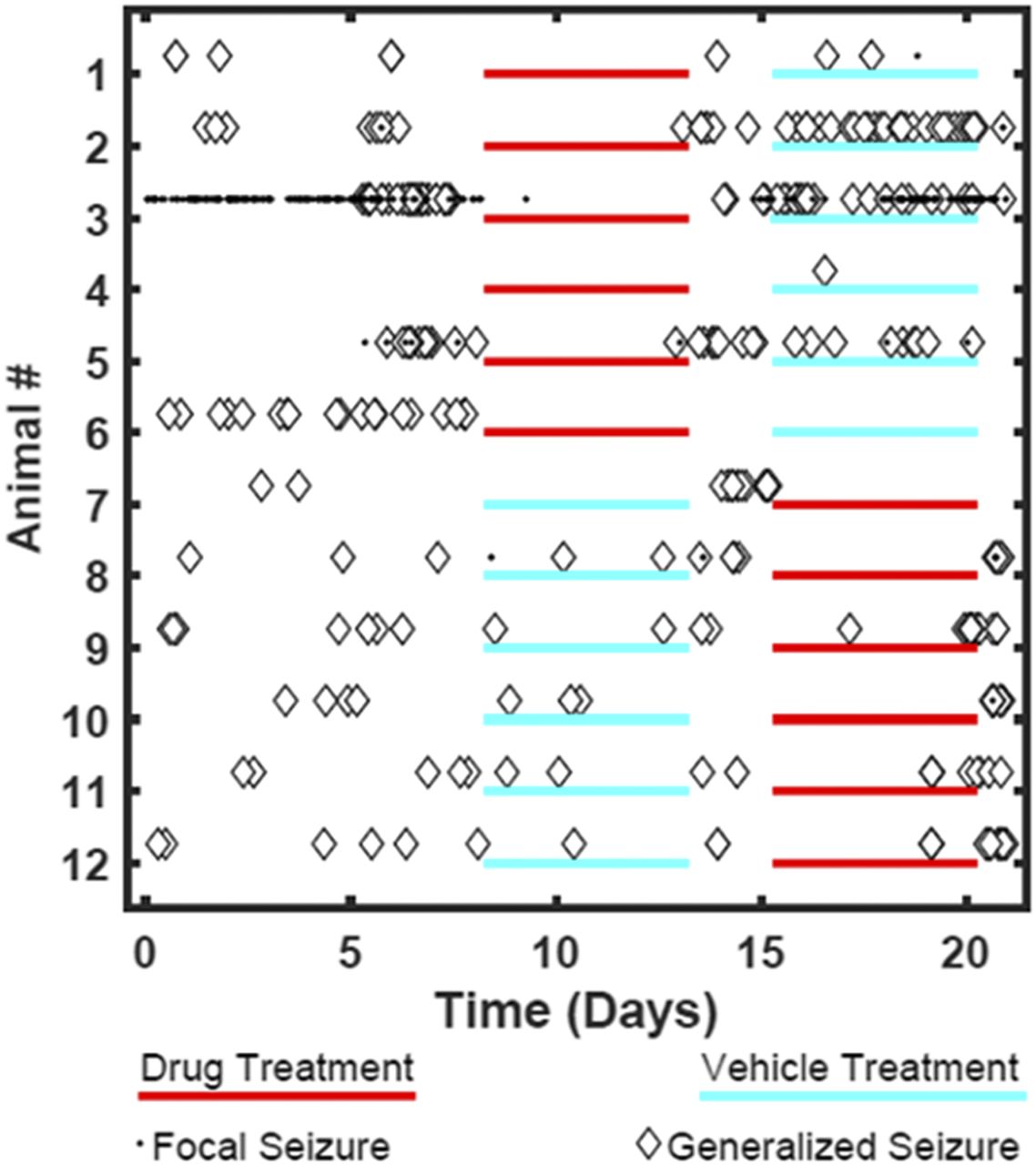
Daily seizure events for each of 12 rats undergoing kainate-induced chronic epilepsy. Data are shown for baseline, vehicle, and drug-treatment conditions. Drug treatment was with KRM-II-81(20 mg/kg, i.p., three times per day).
Effects of KRM-II-81 on seizure outcomes in rats with kainate-initiated chronic epilepsy
Seizure burden data are mean ± S.E.M. for 12 rats. Seizure freedom data are the number of rats that are seizure free/number of rats tested. KRM-II-81 was given three times per day (20 mg/kg, i.p.). *P < 0.05 compared with baseline by Wilcoxon rank sum test (daily seizure burden) or Fisher’s exact probability test (seizure freedom).
Traumatic Brain Injury
To evaluate the effect of KRM-II-81 on inhibiting cortical network activity, we used an in vivo two-photon imaging technique to measure activity changes of cortical layer II/III pyramidal neurons in a controlled cortical impact (CCI) model of traumatic brain injury in Thy1-GCaMP6s transgenic mice. In both sham and CCI-injured mice, activities of fluorescent cells were clearly visible in vivo (Fig. 3B). We first compared activity level between sham-injured mice and CCI mice and found that there were dramatic increases in integrated fluorescence (for measuring intensity of spiking activity of individual neurons) and fraction of active neurons (those that fired at least once during the imaging period) in the CCI group 3 months after brain injury (Fig. 3A) (P < 0.001 and P < 0.01 for integrated fluorescence and fraction of active neurons, respectively, Student’s t test).

KRM-II-81 inhibited cortical neuronal hyperactivity in a CCI model of post-traumatic epilepsy in vivo. (A) In vivo two-photon imaging of cortical neurons in layer II/III showed great increases in integrated fluorescence (left, P < 0.001, Student’s t test) and fraction of active neurons (right, P < 0.01) in mice with CCI injury for 3 months (n = 3 in both groups). (B) Representative images of maximal projections of cortical layer II/III GCaMP6-expressing neurons at baseline and 0.5, 1, 2, and 4 hours after injection of KRM-II-81 in vivo (top row). ΔF/F traces of calcium transients of neurons corresponding to the color-circled neurons are shown on the bottom. Note the dramatic decrease in calcium activity 0.5 hours after injection of KRM-II-81. (C) There were dramatic decreases in integrated fluorescence and fraction of active cells 0.5 hours after drug injection, suggesting a decrease in cortical excitatory activity. One-way ANOVA was followed by Bonferroni test. *P < 0.05; **P < 0.01, ***P < 0.001. n = 3 mice.
Three months after CCI, mouse cortical regions next to the CCI injury site were repeatedly imaged at baseline and 0.5, 1, 2, and 4 hours after intraperitoneal injection of 10 mg/kg KRM-II-81 (356 cells from three CCI mice) (Fig. 3B). At 0.5 hours after drug injection, both the mean integrated fluorescence and fraction of active neurons were dramatically reduced when compared with the baseline level of the mice (one-way ANOVA followed by Bonferroni tests, P < 0.01 for both, n = 3 mice) (Fig. 3C). These values gradually recovered to about baseline levels in about 1 to 2 hours after injection.
Molecular Structural Modeling
Structural docking studies were conducted to elucidate unique structural features of KRM-II-81. Two molecules with low interaction with α1-associated GABAA receptors were employed, 7-ethynyl-1-methyl-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one (QH-II-66) (Huang et al., 1998, 2000) and KRM-II-81 (Poe et al., 2016) (Fig. 4). QH-II-66 was docked in a diazepam (DZP)-bound CryoER structure (6HUP) (Masiulis et al., 2019). Overlay of the bound DZP conformation and best-scored pose of QH-II-66 indicated that both compounds bound similarly in the benzodiazepine binding pocket (Fig. 5A). The ring (A) of DZP and QH-II-66 (as shown in Fig. 4) form a hydrophobic interaction with γ2Phe77 (distance 3.5 Å). The pendant phenyl rings (B) of DZP and QH-II-66 (Fig. 4) were packed in an aromatic box formed by α1Tyr210, α1Tyr160, α1Phe100, α1His102, and α1Phe77. The C-loop α1Ser205 side chain forms a bifurcated hydrogen bond with imine nitrogen and carbonyl oxygen atoms of DZP (2.5 and 2.3 Å, respectively) and QH-II-66 (2.2 and 2.3 Å, respectively) (Fig. 5, B and C). The chlorine atom at C7 of DZP forms a hydrogen bond with the α1His102 side chain (2.6 Å). In contrast, acetylene in place of the chlorine atom at C7 of QH-II-66 did not interact with the α1His102 side chain (Fig. 5C).
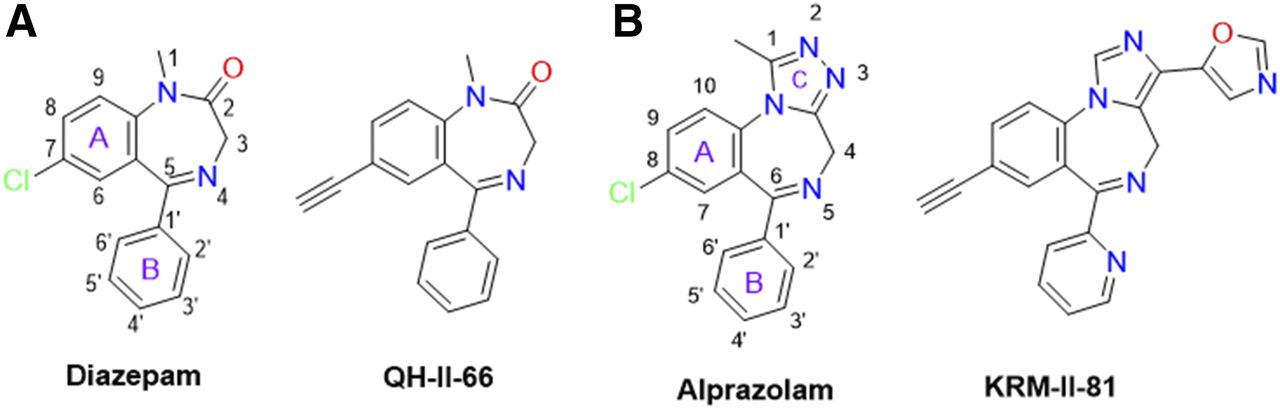
Panel A. Chemical structures of DZP and QH-II-66. The DZP and pendent aromatic rings of DZP are numbered. The benzene (A) and pendent phenyl (B) rings of DZP are labeled. Panel B. Chemical structures of ALP and KRM-II-81. The DZP, pendent aromatic, and triazole rings of ALP are numbered. The benzene (A), pendent phenyl (B), and triazole (C) rings of ALP are labeled.

(A) Bound DZP (orange) and docked QH-II-66 (pink) overlay; α1 (yellow) and γ2 (turquoise) subunits of α1β3γ2L GABAA receptor 6HUP. (B) Ligand-protein interactions; bound DZP (orange) in complex with α1 (yellow) and γ2 (turquoise) subunits of α1β3γ2L GABAA receptor 6HUP. (C) Ligand-protein interactions; docked QH-II-66 (pink) in complex with α1 (yellow) and γ2 (turquoise) subunits of α1β3γ2L GABAA receptor 6HUP. Dashed lines indicate π–π interactions and hydrogen bonds; hydrogen bond (green), hydrophobic interaction (magenta).
KRM-II-81 was docked in the alprazolam (ALP) bound CryoER structure (6HUO). Overlay of the bound ALP conformation and the best-scored pose of KRM-II-81 indicated that the pendant ring (ring B)(Fig. 4) of both compounds bound similarly; however, the diazepine ring (A) and triazole/imidazole rings (Fig. 4) showed slightly different conformations (Fig. 6). The triazole and imidazole rings of ALP and KRM-II-81 formed a hydrophobic π-π interaction with the γ2Tyr58 side chain (distance 4 and 3.9 Å, respectively). The α1Ser205 side chain formed a bifurcated hydrogen bond with the imine nitrogen and triazole ring nitrogen atom of ALP (distance 2.3 and 2.1 Å, respectively). In the case of KRM-II-81, the α1Ser205 side chain can form a similar bifurcated hydrogen bond with the imine nitrogen and imidazole ring nitrogen atoms but with longer distances (2.7 and 2.7 Å, respectively) than observed with ALP. The pendant (B) rings of both ALP and KRM-II-81 (Fig. 4) were packed in an aromatic box formed by α1Tyr210, α1Tyr160, α1Phe100, and α1His102. Ring (A) of ALP (Fig. 4) provided a hydrophobic interaction with γ2Phe77 (3.3 Å) and this interaction was lacking in KRM-II-81. The chlorine atom at C8 of ALP formed a possible halogen bond with the carbonyl oxygen backbone of α1His102 (2.9 Å, angle C8–Cl–O = 167°) (Fig. 7A); this important interaction was lacking in KRM-II-81 (Fig. 7 B).

Bound ALP (gray) and docked KRM-II-81 (sienna) overlay; α1 (yellow) and γ2 (turquoise) subunits of α1β3γ2L GABAA receptor 6HUO.

(A) Ligand-protein interactions; bound ALP (gray) in complex with α1 (yellow) and γ2 (turquoise) subunits of α1β3γ2L GABAA receptor 6HUO. (B) Showing ligand-protein interactions; docked KRM-II-81 (sienna) in complex with α1 (yellow) and γ2 (turquoise) subunits of α1β3γ2L GABAA receptor 6HUO. Dashed lines indicate π–π interactions, hydrogen bonds and halogen bond; hydrogen bond (green), hydrophobic interaction (magenta), and halogen bond (black).
Side-Effect Evaluation.
In rats given 300 mg/kg KRM-II-81, the following reports of sedation, ataxia, or loss of righting were made at each observation time point post dosing: 0.25 hours (0/8 rats); 0.5 hours (0/8 rats); 1 hour (0/8 rats); 2 hours (0/8 rats); 4 hours (0/8 rats); and 6 hours (0/8 rats). Mild behavioral activation in rats was observed at 0.5 and 1 hours postdosing, but these observations were not deemed worthy of note as behavioral toxicity.
Discussion
The need for new antiepileptic drugs continues to be high despite the availability of a host of marketed drugs of different mechanistic classes (Gitto et al., 2010). The lack of seizure control in patients is the primary factor driving this demand (Sinha and Siddiqui, 2011). Increasing inhibitory tone in the central nervous system through enhancement of GABAergic inhibition is a proven mechanism for seizure control (Gitto et al., 2010). However, GABAergic medications also exhibit some liabilities that limit their antiepileptic potential. These drugs can produce cognitive impairment, somnolence, sedation, tolerance, and withdrawal seizures that create dosing limitations such that they are generally used only for acute convulsive episodes (Gitto et al., 2010).
The sedative effects of PAMs of GABAA receptors have been ascribed to their interactions with α1-containing GABAA receptors (McKernan et al., 2000). In addition, it has been shown that agonist activity at α1-containing GABAA receptors, while impacting motor function, is not essential for anticonvulsant efficacy. Genetic inactivation of α1-containing GABAA receptors blocked the motor-impairing effects of diazepam with lesser disruption to its anticonvulsant effects (Rudolph et al., 1999). Furthermore, administration of the α1-selective antagonist β-CCT (Huang et al., 2000), attenuated the motor-impairing effects of diazepam but did not significantly reduce anticonvulsant efficacy (Witkin et al., 2018). Thus, while α1-comprised GABAA receptors mediate sedative and ataxic effects (McKernan et al., 2000), potentiation of α2- and α3-associated GABAA receptors appear to be sufficient for anticonvulsant effects (Rivas et al., 2009; Ralvenius et al., 2015). Therefore, it has been deduced that positive allosteric amplification of α2/3-containing GABAA receptors with selective ligands should provide anticonvulsant efficacy with reduced motor side-effect burden. This hypothesis has been substantiated recently with data from the α2/3-selective GABAA receptor PAM, KRM-II-81, the first selective PAM of α2/3-containing GABAA receptors to be broadly characterized as an anticonvulsant. KRM-II-81 was active in a host of acute and chronic anticonvulsant models in rodents and displayed less motor disruption than the non-α-selective PAM diazepam and less respiratory depression than alprazolam (Witkin et al., 2018). The idea that reduced motor effects of GABAA receptor PAMs could enhance efficacy has also been supported by experimental data: KRM-II-81dispayed broader anticonvulsant efficacy in a series of rodent models and was more efficacious than diazepam in some of the models tested (Witkin et al., 2018).
Although active in multiple rodent anticonvulsant assays (Witkin et al., 2018) of predictive validity (Simonato et al., 2014; Barker-Haliski et al., 2017), additional assurance of the superior anticonvulsant properties of KRM-II-81 was sought in the present set of experiments using models for more advanced stages of the antiepileptic drug screening process (Barker-Haliski et al., 2017). In corneal seizure kindling in mice, KRM-II-81 demonstrated complete seizure blockade and reduction of seizure severity with a minimal effective dose of 15 mg/kg, given orally. The data from this model predict efficacy of KRM-II-81 against secondarily generalized partial seizures in patients (Matagne and Klitgaard, 1998; Barker-Haliski et al., 2017; Hammond et al., 2017).
The potential of KRM-II-81 to impact pharmaco-resistant epileptic patients was also addressed. KRM-II-81 attenuated seizures induced by 6 Hz stimulation (Witkin et al., 2018), a model that is in wide use to predict novel treatments (Barton et al., 2001) and those that might address the needs of pharmaco-resistant epileptic patients (Wilcox et al., 2013; Leclercq et al., 2014). Data from other rodent seizure models used in antiepileptic drug screening programs to guide new compounds in the pharmaco-resistant epilepsy therapy space (Wilcox et al., 2013; Barker-Haliski et al., 2017) were also reported in the present paper. KRM-II-81 suppressed the spontaneous recurrent hippocampal paroxysmal discharges resulting from prior neurotoxic insult by kainate in the mesial temporal lobe epilepsy model. These new preclinical findings predict the efficacy of KRM-II-81 in temporal lobe epilepsy (Bouilleret et al., 1999; Riban et al., 2002). KRM-II-81 was also active in the lamotrigine-resistant kindling model, predicting positive impact on pharmaco-resistant epilepsy (Srivastava et al., 2013; Wilcox et al., 2013; Barker-Haliski et al., 2017). Chronic spontaneous recurrent seizures in epilepsy were modeled in the present study with a repeated low-dose kainate model in rats (Barker-Haliski et al., 2017; Grabenstatter and Dudek, 2019). Clinically relevant measures of human epilepsies with chronic recurrent spontaneous seizures were suppressed by KRM-II-81. KRM-II-81 also significantly increased the percentage of animals that were seizure free in this chronic epilepsy model.
To compare the efficacy of KRM-II-81 to other antiepileptic drugs, data from the rodent assays used in the present experiment are summarized in Table 3. The data demonstrate the efficacy of KRM-II-81 in all models with high potency compared with other antiepileptics. Moreover, the data show that in the pharmaco-resistant rodent models (mesial temporal lobe, lamotrigine-insensitive kindling, and kainite-rendered chronic epileptic rat), multiple standard-of-care antiepileptic drugs did not block seizures, whereas KRM-II-81 was active. In the corneal kindling model, KRM-II-81 was more potent than other drugs that have been studied. Although other GABA modulators were active in the model, topiramate was only half-way effective up to a dose of 59 mg/kg. In the mesial temporal lobe mouse model, KRM-II-81 reduced spontaneous seizures at 15 mg/kg given orally (lower doses were not tested); while levetiracetam was also active in this model (800 mg/kg), lamotrigine, valproate, and carbamazepine were not. In the lamotrigine-insensitive rat kindling model, KRM-II-81 was active against all measures of kindled seizures (severity, prevalence, and duration) at doses from 5 to 20 mg/kg, i.p. In contrast, this model was not responsive to a host of other antiepileptic agents. In rats with chronic epilepsy induced by kainite, KRM-II-81 reduced both seizure burden and the percentage of rats showing seizures. Although some other antiepileptic drugs are also active in this model, they are less potent. Antiepileptics that do not significantly impact seizures in this model of pharmaco-resistant epilepsy are contrasted with that of KRM-II-81 in Table 3.
Comparative efficacy of antiepileptic drugs in rodent seizure models relative to KRM-II-81
In the present report, we also began to explore the possibility that KRM-II-81 might reduce cortical hyperactivity that arises after traumatic brain injury since it is known that TBI is associated with post-traumatic epilepsy (Semple et al., 2019). We used a model of controlled-cortical impact to induce TBI in mice (Ping and Jin, 2016), where the mice have an increased risk of developing post-traumatic epilepsy (Bolkvadze and Pitkänen, 2012). Here, we used imaging analysis to show for the first time that KRM-II-81 (10 mg/kg, i.p.) significantly reduced the heightened cortical excitation in these mice. It is currently thought that antiepileptic medications have little therapeutic impact early after TBI (Wat et al., 2019) and there is even less evidence suggesting efficacy in later phases for reducing post-traumatic epilepsy (Temkin et al., 1990; Thompson et al., 2015). Because of urgent medical need (Temkin, 2009), the potential value of a dampener of TBI-induced cortical excitation in helping TBI patients should be considered. The current findings with cortical excitatory reductions by KRM-II-81 are encouraging of further work.
The α1His102 residue in the benzodiazepine binding pocket plays a key role in the binding of clinically used benzodiazepines (Wieland et al., 1992; Duncalfe et al., 1996; Berezhnoy et al., 2004). The importance of the α1His102 residue has been shown by cysteine crosslinking experiments, in which isothiocyanate substitutions at the C7 position of diazepam reacted with cysteines introduced at α1His102 (Middendorp et al., 2014). Some of the structural features of KRM-II-81 that imbue it with its novel GABAA receptor properties were explored in the present study. Docking studies were performed using ligand bound CryoER structures. The recently published CryoER structure 6HUP (α1β3γ2L GABAA receptor in complex with diazepam) showed that the α1His102 side chain interacts with the chlorine atom at the C7 position of diazepam (Masiulis et al., 2019). Another CryoER structure 6HUO (α1β3γ2L GABAA receptor in complex with alprazolam) showed that the C8 chlorine atom of alprazolam interacts with the α1His102 side chain and can form a halogen bond with the carbonyl oxygen of the α1His102 backbone (Masiulis et al., 2019). Docking results from the present modeling experiments indicated that replacing C7 and C8 chlorine atoms in diazepam and alprazolam, respectively, with acetylene in QH-II-66 and KRM-II-81 leads to a loss in this key interaction with α1His102. These data provide structural rationale for why QH-II-66 (Huang et al., 1998, 2000) and KRM-II-81 (Poe et al., 2016) have low affinity toward α1-containing GABAA receptors and for the lower liability of motor impairment (associated with agonism at α1-containing GABAA receptors) (McKernan et al., 2000) observed with KRM-II-81 compared with diazepam (Witkin et al., 2018). This decreased interaction with α1His102 might also create reductions in other liability characteristics of nonselective GABAA receptor PAMs like diazepam (e.g., tolerance and dependence, as discussed previously). Indeed, in the present study, 300 mg/kg KRM-II-81 was studied at 0.25–6 hours postdosing without any observable sedation, ataxia, or loss of righting. This dose is >10-fold higher than the doses that are active in the rodent assays detecting anticonvulsant activity. The findings support the previously reported differences in effects of diazepam or alprazolam compared to KRM-II-81 (Witkin et al., 2018). The relatively milder impact of KRM-II-81 on motor function is congruent with the molecular modeling data provided in the present report.
In addition to the rodent model–based evidence presented here and elsewhere (Witkin et al., 2018), KRM-II-81 also has efficacy in human epileptic tissue. In cortical tissue freshly transected from juvenile epileptic patients, KRM-II-81 attenuated the firing rates of neurons in tissue slices in a microelectrode array (Witkin et al., 2018, 2019b). Human data also have helped to substantiate the proposition of reduced motor burden with allosteric potentiators with selectivity for α2/3 over α1 protein assemblies (Zuiker et al., 2016). Recent studies with another ligand are also at a stage where human data have been presented. PF‐06372865 (7-ethyl-4-(4'-(ethylsulfonyl)-6-fluoro-2'-methoxy-[1,1'-biphenyl]-3-yl)-7H-imidazo[4,5-c]pyridazine) is selective for α2/3/5- versus α1-associated GABAA receptors (Nickolls et al., 2018) and has been reported to block pentylenetetrazol and amygdala kindled seizures in rodents (Buhl et al., 2017 cited in Duveau et al., 2019) and in the Genetic Absence Epilepsy Rat from Strasbourg (GAERS) rat, a genetic model of absence seizures (Duveau et al., 2019). PF‐06372865 was studied in healthy human volunteers and in patients where it was well tolerated (Gurrell et al., 2019; Nickolls et al., 2018; Simen et al., 2019). Most importantly for the present purposes, PF‐06372865 was efficacious in inhibiting electrical activity in patients with photosensitive epilepsy in a provocation model (). The photosensitive epilepsy model is gaining use as a model with good predictive validity (Kasteleijn-Nolst Trenité et al., 1996; French et al., 2014; Yuen and Sims, 2014).
In conclusion, an opportunity exists for the development of novel antiepileptic drugs based on their selectivity for α2/3-associated versus α1-associated GABAA receptor PAMs. Thus, KRM-II-81 is predicted to provide broad efficacy across a range of epileptic conditions, provide enhanced efficacy compared with nonselective GABAA receptor PAMs, and importantly to impact epileptic phenomenon in pharmaco-resistant patients. There are data to suggest that these selective PAMs might also have less liability than their nonselective relatives to produce amnesia, tolerance, or abuse (Ator et al., 2010; Atack, 2011; Ralvenius et al., 2015; Zuiker et al., 2016; Moerke et al., 2019). Thus, the imidizodiazepine, KRM-II-81, demonstrates reduced motor impairment, respiratory depression, and abuse liability compared with benzodiazepines such as diazepam or alprazolam (Witkin et al., 2018; Moerke et al., 2019). An additional benefit from KRM-II-81 is enabled by its ability to provide relief of comorbid anxiety (Poe et al., 2016; Witkin et al., 2017) and neuropathic pain (Witkin et al., 2019a).
Acknowledgments
This paper is dedicated to Dr. Werner Sieghart for his pioneering work on the GABA system and rigorous approach to neuroscience. We thank the National Institutes of Health Anticonvulsant Screening Program for data collection in the epilepsy models. We are also grateful to John and Nancy Peterson for their support of this research. Thanks are especially given to the Epilepsy Therapy Screening Program (ETSP) for guidance on this molecule through the testing battery; the valuable work done at the expert laboratories, which is an integral part of this testing consortium; and Dr. Karen Wilcox for sharing data on some of these animal models.
Authorship Contributions
Participated in research design: Witkin, Golani, Xiong, Smith, Cerne, Cook, Jin.
Conducted experiments: Witkin, Golani, Xiong.
Contributed new reagents or analytic tools: Li, Golani, Xiong, Ping, Rashid, Jahan, Cook, Jin.
Performed data analysis: Witkin, Xiong, Jin.
Wrote or contributed to the writing of the manuscript: Witkin, Li, Golani, Xiong, Smith, Ping, Rashid, Jahan, Cerne, Cook, Jin.
Footnotes
- Received July 13, 2019.
- Accepted October 17, 2019.
This work was supported by the Henry and Nellie Pence family for supporting this work through a Pence Foundation Grant (awarded to J.L.S.); the National Institutes of Health [Grants MH-096463 and NS-076517]; the National Science Foundation, Division of Chemistry [Grant CHE-1625735]; the University of Wisconsin-Milwaukee’s Shimadzu Laboratory for Advanced and Applied Analytical Chemistry; the Milwaukee Institute of Drug Discovery; and the University of Wisconsin-Milwaukee Research Foundation.
Some of the authors are patent holders for KRM-II-81 (G.L., L.K.G., F.R., R.J., and J.M.C.). The University of Wisconsin-Milwaukee is the owner of KRM-II-81. J.M.W. is an adjunct faculty member of the University of Wisconsin-Milwaukee and lead biologist on the antiepileptic drug development effort for KRM-II-81.
Abbreviations
- ALP
- alprazolam
- CCI
- controlled cortical impact
- DZP
- diazepam
- EEG
- electroencephalogram
- F
- fluorescence
- KRM-II-81
- 5-(8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo[1,5-a][1,4]diazepin-3-yl)oxazole
- PAM
- positive allosteric modulator PF‐06372865 7-ethyl-4-(4'-(ethylsulfonyl)-6-fluoro-2'-methoxy-[1,1'-biphenyl]-3-yl)-7H-imidazo[4,5-c]pyridazine
- PDB
- Protein Data Bank
- QH-II-66
- 7-ethynyl-1-methyl-5-phenyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one
- TBI
- traumatic brain injury
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}