


Abstract
In the normal heart, frequently used anesthetics such as isoflurane and propofol can reduce inotropy. However, the impact of these agents on the failing myocardium is unclear. Here, we examined whether and how isoflurane and propofol influence cardiac contractility in intact cardiac muscles from rats treated with monocrotaline to induce heart failure. We measured force and intracellular Ca2+ ([Ca2+]i) in trabeculae from the right ventricles of the rats in the absence or presence of propofol or isoflurane. At low to moderate concentrations, both propofol and isoflurane dose-dependently depressed cardiac force generation in failing trabeculae without altering [Ca2+]i. At high doses, propofol (but not isoflurane) also decreased amplitude of [Ca2+]i transients. During steady-state activation, both propofol and isoflurane impaired maximal Ca2+-activated force (Fmax) while increasing the amount of [Ca2+]i required for 50% of maximal activation (Ca50). These events occurred without apparent change in the Hill coefficient, suggesting no impairment of cooperativity. Exposing these same muscles to the anesthetics after fiber skinning resulted in a similar decrement in Fmax and rise in Ca50 but no change in the myofibrillar ATPase-Ca2+ relationship. Thus, our study demonstrates that challenging the failing myocardium with commonly used anesthetic agents such as propofol and isoflurane leads to reduced force development as a result of lowered myofilament responsiveness to Ca2+.
SIGNIFICANCE STATEMENT Commonly used anesthetics such as isoflurane and propofol can impair myocardial contractility in subjects with heart failure by lowering myofilament responsiveness to Ca2+. High doses of propofol can also reduce the overall amplitude of the intracellular Ca2+ transient. These findings may have important implications for the safety and quality of intra- and perioperative care of patients with heart failure and other cardiac disorders.
Introduction
Since the very first demonstration made by Dr. William T. Morton at the Massachusetts General Hospital in October 1846, general anesthesia has become an inseparable part of patient procedures (both surgical and nonsurgical). However, it is increasingly evident that anesthetics may have a wide array of off-pathway (side) effects, especially in patients with severe disease conditions and comorbidities (Martin, 2005; Reich et al., 2005; Armstrong et al., 2017; Kawasaki et al., 2018; Juhász et al., 2019). For anesthesiologists, of particular concern are patients with heart failure (HF) (Smit-Fun and Buhre, 2016). In the last few decades, the number of procedures that have become amenable to individuals with moderately depressed heart function has increased (Benjamin et al., 2017). Some common procedures may include cardiac-related operations, such as implantation of ventricular assist devices, pacemakers, synchronized stimulators, and defibrillators, and noncardiac procedures from hip replacement to endoscopies. However, recent clinical studies have shown that the perioperative mortality and morbidity in HF patients remain high (Flu et al., 2010; Mebazaa et al., 2010), as much as 2- to 3-fold higher than for patients with coronary artery disease (Hernandez et al., 2004; Hammill et al., 2008). These high rates of morbidity and mortality result from complications such as heart attack, worsening of HF, and other organ failure (Lee et al., 1999; Poldermans et al., 2009).
It has been well documented that anesthetic agents affect myocardial contraction in normal myocardium (Bosnjak et al., 1992; Hebbar et al., 1997; Jiang and Julian, 1998; Davies et al., 2000; Sprung et al., 2001; Bovill, 2006). Moreover, we have shown that the inhalational agent isoflurane, at low-to-moderate doses (0.5%–1.5%), reduces myofilament Ca2+ responsiveness in normal rat trabeculae without altering intracellular Ca2+ levels (Ding et al., 2011). We also demonstrated that propofol jeopardizes force development in healthy rat cardiac muscle via decreased myofilament Ca2+ responsiveness (Meng et al., 2016). Furthermore, we have reported that both propofol and isoflurane produce a direct effect on myofilament function, and we have identified several potential key targets for these anesthetics within the contractile proteins (Meng et al., 2016). Nevertheless, the impact of anesthetic agents on cardiac force generation in failing preparations remains poorly characterized, particularly from a mechanistic point of view, and is somewhat controversial. Indeed, studies have shown that inhalational agents can depress myocardial contractility in both failing and healthy hearts (Pagel et al., 1996; Preckel et al., 2004). However, other reports suggest that the failing myocardium can be relatively insensitive to inhalational agents (Kissin et al., 1983; Vivien et al., 1997). Of note, in studies that have demonstrated a reduction in cardiac function by anesthetics, the mechanism of impairment has not been clarified.
In the present study, we tested the hypothesis that both isoflurane and propofol (two widely used anesthetic agents) depress force in the failing myocardium, and examined whether the negative inotropic effect stems from a direct action of the agents on the myofilaments.
Materials and Methods
Animal Care.
A total of 36 LBN/F1 rats of either sex (250–300 g; Harlan Laboratories, Indianapolis, IN) were used in these experiments. Animal care and experimental protocols were approved by the Animal Care and Use Committee of the Johns Hopkins University School of Medicine.
Right Heart Failure in the Rat.
Rats received a single intraperitoneal injection of monocrotaline (MCT; 60 mg/kg in saline, MCT group, n = 20) or an equivalent volume of saline (control group, n = 16). No anesthesia was required given the short duration of injection (<5–10 seconds). They were weighed weekly for 3 weeks post injection and then daily. Animals were also monitored for clinical signs of HF, which included successive days of weight loss, dyspnea, cold extremities, or lethargy. Two rats in the MCT group did not develop right heart failure after injection, and two rats died accidentally. These rats were excluded from the study. The experimental matrix is detailed in Supplemental Table 1.
Cardiac function of the rats was evaluated by echocardiography and hemodynamic measurements. Rats from the two groups were lightly sedated with isoflurane (<1%) before undergoing electrocardiogram analysis and temperature monitoring. Then, right ventricular (RV) function was evaluated by echocardiography (Agilent Sonos 5500, 15-MHz probe, 3-cm depth; Agilent Technologies). RV wall thickness and chamber dimensions were measured at the midlevel parasternal long-axis view and parasternal short-axis view, respectively, at the level of the papillary muscles. RV fractional area of change (FAC) was calculated based on manually traced areas of the right ventricle cavity endocardium at the parasternal long-axis view during systole and diastole (i.e., apical four-chamber view) using the following equation: FAC = (end-diastolic area − end-systolic area)/end-diastolic area × 100%. Software associated with the echo machine analyzed the traced areas to derive volumes and calculate the RV ejection fraction. Cardiac output was calculated based on pulse-Doppler wave velocity wave form across the pulmonary valve (i.e., velocity-time integral) and the cross-sectional area of the pulmonary artery: cardiac output = cross-sectional area × velocity-time integral × heart rate.
Determination of Force and Intracellular Ca2+ in Intact Trabeculae.
When the MCT-injected rats exhibited overt HF, they were anesthetized with an intra-abdominal injection of pentobarbital (100 mg/kg). Then, the heart was exposed by midsternotomy, rapidly excised, and placed in a dissection dish. The aorta was cannulated and the heart perfused in a retrograde fashion with dissecting Krebs-Henseleit (K-H) solution equilibrated with 95% O2 and 5% CO2. The dissecting K-H solution was composed of (in millimolar concentrations) NaCl 120, NaHCO3 20, KCl 5, MgCl 1.2, glucose 10, CaCl2 0.5, and 2,3-butanedione monoxime 20 [pH 7.35–7.45 at room temperature (21–22°C)]. Trabecular muscle from the right ventricle of the heart was dissected and mounted between a force transducer and a motor arm, superfused with K-H solution at a rate of ∼10 ml/min, and stimulated at 0.5 Hz.
Force was measured by a force transducer system (KG7; Scientific Instruments GmbH, Heidelberg, Germany) and expressed in millinewtons per square millimeter of cross-sectional area. The muscles underwent isometric contractions with the resting muscle length set so that resting force was 15% of total force development (i.e., optimal muscle length). This resting muscle length, which corresponded to a resting sarcomere length of 2.20–2.30 μm as determined by laser diffraction (Gao et al., 1996), was maintained throughout the experiments.
Intracellular Ca2+ ([Ca2+]i) was measured by using the free acid form of fura-2 (Dai et al., 2007; Gao et al., 2012). Fura-2 potassium salt was microinjected iontophoretically into one cell and allowed to spread throughout the whole muscle (via gap junctions) without affecting force development. The tip of the electrode (∼0.2 μm in diameter) was filled with fura-2 salt (1 mM), and the remainder of the electrode was filled with 150 mM KCl. After successful impalement into a superficial cell in nonstimulated muscle, a hyperpolarizing current of 5–10 nA was passed continuously for ∼15 minutes. In some preparations, multiple injection sites were needed to achieve the desired loading of fura-2 because the duration of each injection was limited to <10 minutes. Fura-2 epifluorescence was measured by excitation at 380 and 340 nm. Fluorescent light was collected at 510 nm by a photomultiplier tube (R1527; Hamamatsu Photonics, Hamamatsu, Japan). The output of the photomultiplier was collected and digitized. [Ca2+]i was given by the following equation (after subtraction of the autofluorescence): (1)where R is the observed ratio of fluorescence (340/380 nm), K′d is the apparent dissociation constant, Rmax is the ratio of 340 nm/380 nm at saturating Ca2+ concentration, and Rmin is the ratio of 340/380 nm at 0 Ca2+ concentration. The values of K′d, Rmax, and Rmin were determined by in vivo calibrations as described previously (Gao et al., 1994, 1998).
(1)where R is the observed ratio of fluorescence (340/380 nm), K′d is the apparent dissociation constant, Rmax is the ratio of 340 nm/380 nm at saturating Ca2+ concentration, and Rmin is the ratio of 340/380 nm at 0 Ca2+ concentration. The values of K′d, Rmax, and Rmin were determined by in vivo calibrations as described previously (Gao et al., 1994, 1998).
Ryanodine (1.0 μM) was used to enable steady-state activation. After the muscles were exposed to ryanodine for 15 minutes, different levels of tetanization were induced briefly (∼4–8 seconds) by stimulating the muscles at 10 Hz at various concentrations of extracellular Ca2+ concentrations (0.5–20 mM). The steady-state force–[Ca2+]i relations were fit with a function of the Hill equation: (2)where F is the steady-state force at various [Ca2+]i, Fmax is the maximal Ca2+-activated force, Ca50 is the [Ca2+]i required to achieve 50% maximal activation, and n is the Hill coefficient.
(2)where F is the steady-state force at various [Ca2+]i, Fmax is the maximal Ca2+-activated force, Ca50 is the [Ca2+]i required to achieve 50% maximal activation, and n is the Hill coefficient.
Chemical Skinning of Trabecular Muscles.
After the steady-state experiments, the same trabeculae were immediately skinned by 15–20 minutes of exposure to Triton X-100 (1%) in relaxing solution containing (in millimolar concentrations) KCl 100, HEPES 25, K2EGTA 10, creatine phosphate sodium salt (Na2CrP) 15, Na2ATP 5, MgCl2 5.15, and leupeptin 0.5 (pH 7.2 with KOH). Activating solution contained (in millimolar concentrations) Ca2+-EGTA 10, KCl 100, HEPES 25, Na2CrP 15, Na2ATP 5, MgCl2 4.75, and leupeptin 0.5 (pH 7.2). Different Ca2+ concentrations were obtained by mixing the activating solution and relaxing solution in different ratios. The readiness of the skinned preparation was confirmed by the loss of pink color, clear visibility of the sarcomeres, and instantaneous force development upon exposure to activating solutions. Diastolic sarcomere length was determined by direct visualization under 100× magnification and was set at ∼2.2 μm. Resting force was usually 10%–15% of maximal activated force at this sarcomere length. Force–Ca2+ relations were obtained by exposing the skinned muscles to activating solutions with various extracellular Ca2+ concentrations and were fit with the Hill equation: (3)where F is the steady-state force at various [Ca2+], Fmax is the maximal Ca2+-activated force, Ca50 is the [Ca2+] required to achieve 50% maximal activation, and n is the Hill coefficient.
(3)where F is the steady-state force at various [Ca2+], Fmax is the maximal Ca2+-activated force, Ca50 is the [Ca2+] required to achieve 50% maximal activation, and n is the Hill coefficient.
Determination of Myofibrillar ATPase Activity.
Myofibrils were prepared from the failing right ventricle as previously described (Murphy and Solaro, 1990; Dai et al., 2007) with careful use of protease inhibitors. Assays were carried out under incubation conditions established by varying the total concentration of metals, salts, and ligands; ionic strength was maintained by using stability constants compiled by Fabiato (1981). Assays were performed at pH 7.0 with 50 mM imidazole, 50 mM KCl, and 2 mM MgATP. Inorganic phosphate liberation was measured by using a microtiter plate version of the standard assay as described by Rarick et al. (1997). Protein concentration was determined by a variation of the Lowry method (Bio-Rad Laboratories, Hercules, CA). In the final assay conditions, the myofibrillar protein concentration was diluted in buffer to a concentration of 0.2 mg/ml; the protein concentration was determined again for final calculations. Mg-ATPase activity was calculated in nanomoles of inorganic phosphate liberated per milligram of myofibrillar protein per minute.
Preparation of Isoflurane and Propofol.
Isoflurane was purchased from Baxter Healthcare Corporation (Deerfield, IL) and dissolved into working solutions as a percentage by isoflurane vaporizer with O2 as a carrier. The concentrations of isoflurane were expressed as percentages. We checked the concentration of isoflurane in the experimental solution periodically using high-performance liquid chromatography to ensure that the desired concentrations were delivered. Propofol was purchased from Sigma-Aldrich (St. Louis, MO) in powder form and dissolved in DMSO as a stock solution. The final concentration of DMSO at the desired working propofol concentrations was less than 0.1%. In preliminary studies, we determined that this DMSO concentration had no effect on force development or [Ca2+]i transients (data not shown).
Statistical Analysis.
We determined the number of samples/experiments based on a probability for α < 0.05, a power of 0.80, and an anticipated depression in force development of >20% of baseline or control value with S.E.M. = 6%–8%. Paired t test and one-way ANOVA were used for statistical analysis of the data (Systat 10.2.01; Systat Software Inc., San Jose, CA). A value of P < 0.05 was considered to indicate statistically significant differences between groups. Unless otherwise indicated, pooled data are expressed as means ± S.E.M.
Results
Both Isoflurane and Propofol Decrease Force Development in Trabecular Muscles from Nonfailing Hearts Without Affecting Amplitude of the [Ca2+]i Transient.
We first evaluated the effect of isoflurane and propofol on contraction in isolated trabecular muscles from the nonfailing hearts of saline-injected rats (Table 1). Figure 1 shows the pooled data of all muscles exposed to isoflurane or propofol. Isoflurane significantly reduced developed force (starting at 1%, P < 0.05) but did not alter [Ca2+]i transient amplitudes at the concentrations tested (P > 0.05; Fig. 1A). Propofol significantly diminished developed force beginning at 30 μM and reduced [Ca2+]i amplitude when the concentration reached 100 μM (Fig. 1C). The steady-state force–[Ca2+]i relation obtained before and after anesthetic exposure showed that both anesthetics decreased Fmax. With isoflurane, Fmax decreased from 80 ± 6 to 58 ± 5 mN/mm2 (P < 0.05), and with propofol, Fmax decreased from 84 ± 4.3 to 49.6 ± 4.7 mN/mm2 (P < 0.05). Both agents increased the Ca50. With isoflurane, Ca50 increased from 0.526 ± 0.038 to 1.213 ± 0.085 μM (P < 0.05), whereas with propofol, it increased from 0.56 ± 0.08 to 0.74 ± 0.04 μM (P < 0.05; Fig. 1, B and D). Neither isoflurane nor propofol significantly affected the Hill coefficient. This parameter changed from 4.11 ± 0.36 to 2.29 ± 0.19 (P < 0.05) after isoflurane treatment and from 4.8 ± 0.37 to 2.98 ± 0.39 after propofol (P < 0.05). We did not observe any difference between muscles from either sex. Consistent with previous findings (Ding et al., 2011; Meng et al., 2016), these data suggest that in healthy cardiac muscles, the primary targets of isoflurane and propofol are the myofilaments.
Cardiac performance indices of right ventricle in control and monocrotaline-injected rats

Effect of isoflurane and propofol on contraction and [Ca2+]i transients in normal cardiac muscle. (A) Pooled data of trabecular force development and amplitudes of intracellular Ca2+ transients in the presence of different concentrations of isoflurane. Force decreased in a dose-dependent manner as concentrations of isoflurane increased, but amplitudes of [Ca2+]i transients remained unchanged (n = 8). (B) Steady-state force–[Ca2+]i relations in the presence and absence of isoflurane (1.5%) in isolated intact trabecular muscles. The steady-state forces were plotted against amplitudes of [Ca2+]i transients, which were grouped into bins of 0.5–1 µM ranges (n = 6). (C) Pooled data of trabecular force development and amplitudes of intracellular Ca2+ transients in the presence of different concentrations of propofol. Force decreased in a dose-dependent manner as concentrations of propofol increased. Amplitudes of [Ca2+]i transients remained unchanged until propofol concentrations reached 100 μM (n = 8). (D) Steady-state force–intracellular Ca2+ relations in the presence and absence of propofol (50 µM) in isolated intact cardiac trabecular muscles. The steady-state forces were plotted against amplitudes of [Ca2+]i transients, which were grouped into bins of 0.5–1 µM ranges (n = 6). *P < 0.05 vs. baseline, paired t test, temperature = 22°C.
A Single Dose of MCT Induces RV Remodeling and Dysfunction.
Next, we sought to determine the effect produced by these two anesthetic agents on intact cardiac muscles from failing rat hearts. To this end, we induced RV failure by administering one dose (60 mg/kg) of MCT intra-abdominally, which has been shown to induce pulmonary hypertension followed by RV hypertrophy and failure (Kögler et al., 2003; Stones et al., 2013). Consistent with these previous findings, here we found that 4 to 5 weeks after MCT injection, rats exhibited signs of RV failure—namely, low food intake, retarded weight gain, and lack of daily activity—as compared with saline-injected rats. Echocardiography revealed markedly decreased RV contractility as indicated by smaller ejection fraction, less FAC, and less cardiac output. The RV was also manifestly dilated (Fig. 2; Table 1), denoting severe RV remodeling. There were no differences in response to MCT injection between male and female rats.

A representative transthoracic echocardiographic image [short axis transverse view (SAX)] of a rat heart showing a severely enlarged right ventricle with flattening of the ventricular septum (SEP). The image was taken 4.5 weeks after the rat received an intra-abdominal injection of monocrotaline (60 mg/kg). The rat also showed signs of right heart failure at the time of the echocardiograph: inactivity, labored breathing, significant loss of body weight, and enlarged liver. BPM, beats per minute; LV, left ventricle.
Isoflurane and Propofol Both Reduce Twitch Force but with Differing Effects on [Ca2+]i Transients.
Whether isoflurane or propofol alters force development and whole [Ca2+]i transients in isolated (intact) failing trabecular muscle preparations is currently unclear. To fill this gap in knowledge, we administered isoflurane or propofol to intact (RV) trabeculae isolated from MCT-injected rats. Figure 3 shows the typical raw recordings of force and [Ca2+]i transients at different isoflurane concentrations and the pooled data of all muscles tested. Isoflurane weakened force development in a dose-dependent manner without significantly altering [Ca2+]i transient amplitude at any concentration tested (up to 3%). Force was reduced significantly at isoflurane concentrations >1.5%.

Effect of isoflurane on force and [Ca2+]i transient in failing rat cardiac muscle. (A) Raw recordings of force development (left panel) and corresponding [Ca2+]i transients (right panel) of a trabecular muscle in the presence of different isoflurane concentrations. The muscle was stimulated at 0.5 Hz in the presence of 1.0 mM external Ca2+ at room temperature (22°C). (B) Pooled data of trabecular force development and amplitudes of [Ca2+]i transients in the presence of different isoflurane concentrations. Force development decreased in a dose-dependent manner as concentrations of isoflurane increased. Force development became significantly less than that at baseline at concentrations greater than 1.5%. Amplitudes of [Ca2+]i transients remained unchanged at all concentrations tested. Temperature = 22°C; external Ca2+ = 1.0 mM; stimulation rate = 0.5 Hz. *P < 0.001; n = 8.
At concentrations between 0 and 30 μM, propofol did not significantly alter either developed force or [Ca2+]i; however, at 40 μM and higher, it reduced force development. Only at concentrations above 50 μM did it also significantly decreased [Ca2+]i transient (Fig. 4). In essence, isoflurane decreases force development by decreasing myofilament Ca2+ responsiveness in failing RV muscle. In contrast, propofol has a dual negative effect on Ca2+-dependent cardiac force generation: at low concentrations, the reduction in force stems from changes in myofilament properties (i.e., decreased myofilament Ca2+ responsiveness), whereas at moderate/high doses, this decrement originates from decreased Ca2+ cycling/availability.

Effect of propofol on force and [Ca2+]i transient in failing rat cardiac muscle. (A) Raw recordings of force development (left panel) and corresponding [Ca2+]i transients (right panel) of a trabecular muscle in the presence of different propofol concentrations. The muscle was stimulated at 0.5 Hz in the presence of 1.0 mM external Ca2+ at room temperature (22°C). (B) Pooled data of trabecular force development and amplitudes of [Ca2+]i transients in the presence of different propofol concentrations. Force development decreased in a dose-dependent manner as concentrations of propofol increased. Force development became significantly less than that at baseline at concentrations of 40 µM and higher. Amplitudes of [Ca2+]i transients remained unchanged at all concentrations tested. Temperature = 22°C; external Ca2+ = 1.0 mM; stimulation rate = 0.5 Hz. *P < 0.001; n = 8.
Effect of Propofol and Isoflurane on Myofilament Ca2+ Responsiveness.
We next obtained steady-state force–[Ca2+]i relationships to directly assess the myofilament properties that underlie changes in myofilament Ca2+ responsiveness. We determined this relationship in the failing RV muscle before and during anesthetic exposure. At baseline, the steady-state force–[Ca2+]i relationship was diminished in failing trabecular muscles as compared with that in nonfailing muscles. Exposure to 1.5% isoflurane also significantly lowered Fmax from 37.89 ± 4.45 to 22.97 ± 2.1 mN/mm2 (P < 0.05 vs. before isoflurane). Yet the Ca50 was unchanged (0.65 ± 0.11 and 0.5 ± 0.06 μM, respectively; P > 0.05; Fig. 5A; Table 2). Likewise, propofol (50 μM) reduced Fmax from 32.47 ± 3.23 to 22.48 ± 1.7 mN/mm2 (P < 0.05) but did not alter Ca50 (0.5 ± 0.08 and 0.65 ± 0.1 μM, respectively; P > 0.05; Fig. 6A; Table 2). Notably, neither propofol nor isoflurane affected the Hill coefficient (with isoflurane: 2.15 ± 0.26 vs. without isoflurane: 2.81 ± 0.18, P > 0.05; with propofol: 2.5 ± 0.14 vs. without propofol: 4.27 ± 0.12, P > 0.05). This set of data shows that propofol and isoflurane primarily decreased Fmax in the intact muscle.
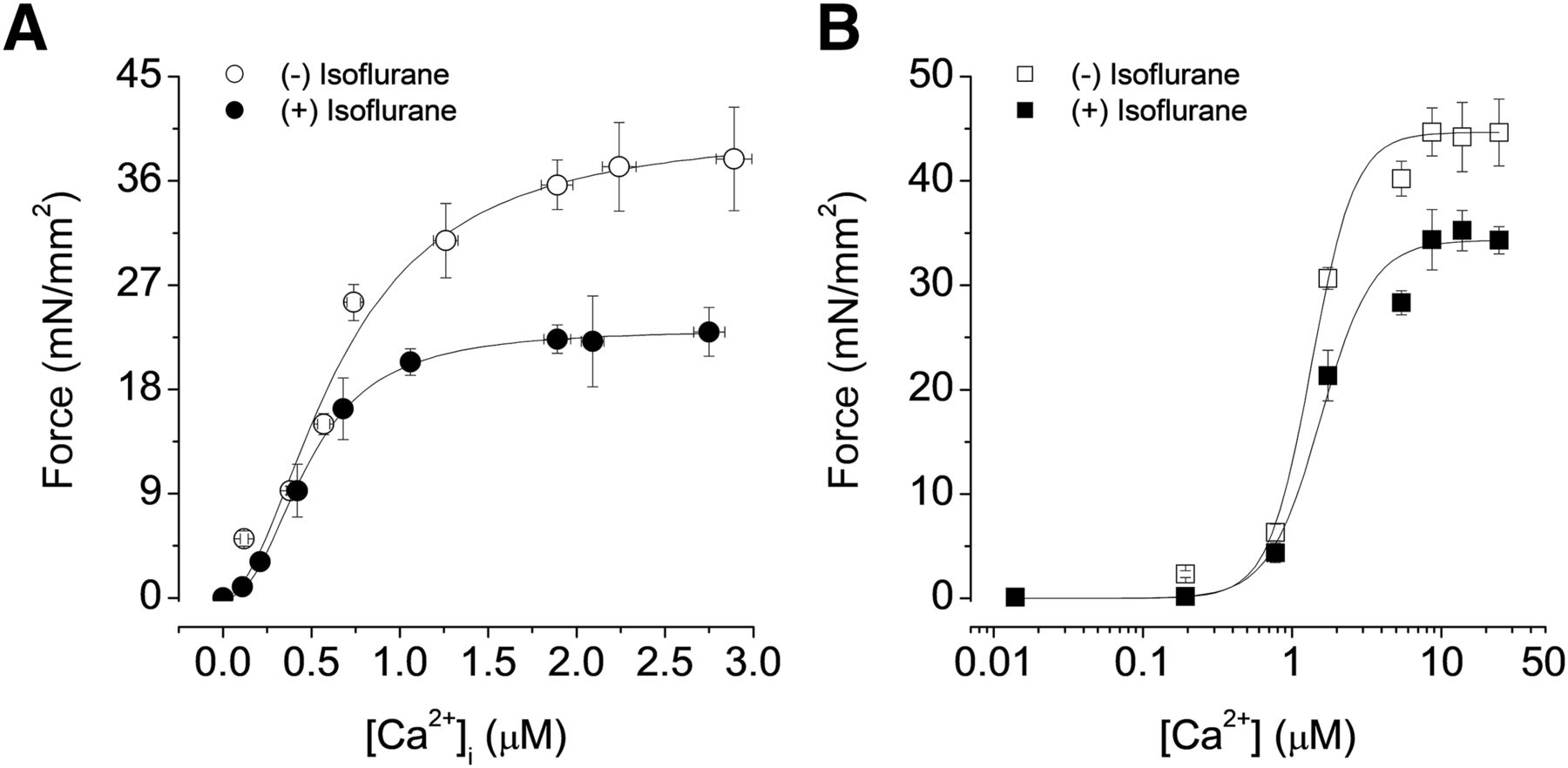
Steady-state force–[Ca2+]i relations in the presence and absence of isoflurane (1.5%) in isolated intact and skinned failing trabecular muscles. The steady-state forces were plotted against amplitudes of [Ca2+]i transients, which were grouped into 0.5-µM bins. (A) In intact trabeculae, Fmax was significantly reduced and Ca50 was significantly increased by isoflurane. (B) The same muscles in which steady-state relations were first obtained were chemically skinned and activated with various Ca2+ concentrations. The effect of isoflurane on Fmax and Ca50 persisted in these skinned muscles. Temperature = 22°C; n = 6.
Parameters for force–Ca2+ relationships in intact and skinned failing cardiac trabecular muscles

Steady-state force–[Ca2+]i relations in the presence and absence of propofol (50 μM) in isolated intact and skinned failing trabecular muscles. The steady-state forces were plotted against amplitudes of [Ca2+]i transients, which were grouped into 0.5-μM bins. (A) In intact trabeculae, Fmax was significantly reduced and Ca50 was significantly increased by propofol. (B) The same muscles in which steady-state relations were first obtained were chemically skinned and activated with various Ca2+ concentrations. The effect of propofol on Fmax and Ca50 persisted in these skinned muscles. Temperature = 22°C; n = 6.
We next determined the steady-state force–[Ca2+]i relations in skinned muscles to rule out cytosolic factors that could potentially influence this relationship (Gao et al., 1994). In the presence of isoflurane, Fmax was significantly depressed from 44.66 ± 3.21 to 34.32 ± 1.3 mN/mm2 (P < 0.05) in skinned fibers. Ca50 was unaffected (1.35 ± 0.15 and 1.55 ± 0.18 µM, respectively; P > 0.05; Fig. 5B; Table 2). Similarly, in skinned preparations, propofol (50 μM) markedly reduced Fmax from 42.42 ± 3.31 to 33.36 ± 2.02 mN/mm2 (P < 0.05), again without affecting Ca50 (1.3 ± 0.15 and 1.56 ± 0.25 µM, pre- and postanesthetic, respectively; P > 0.05; Fig. 6; Table 2). These data, indicating that isoflurane- and propofol-induced depression of the steady-state force–Ca2+ relation persists in skinned muscles, lend further support to the contention that these agents affect myocardial contraction primarily via a direct effect on the myofilament status.
Neither Propofol nor Isoflurane Alter Myofibrillar Mg-ATPase Activity.
Next, we determined the Mg-ATPase activity in isolated myofibrils from failing right ventricles treated with propofol or isoflurane to examine whether the anesthetics also affect the energetics of contraction itself. We observed that the overall maximal ATPase activity was diminished in the failing myocardium (55.18 ± 1.38 nM Pi/mg protein per minute; Fig. 7), consistent with previously reported findings (LeWinter, 2005; Joubert et al., 2008). However, neither propofol nor isoflurane affected the Mg-ATPase–pCa relationship (Fig. 7). In fact, the maximal Ca2+-activated Mg-ATPase was 57.92 ± 3.7 nM Pi/mg protein per minute in the presence of isoflurane and 54.56 ± 1.61 nM Pi/mg protein per minute in the presence of propofol (P > 0.05 vs. failing control; Fig. 7). Moreover, the Ca2+ concentration in the solution for half maximally activated Mg-ATPase activity was 6.31 ± 0.21 μM for the failing preparation at baseline, and 6.33 ± 0.34 and 6.29 ± 0.31 μM after propofol and isoflurane, respectively (P > 0.1). Thus, these anesthetic agents decrease force generation without affecting cross-bridge ATP consumption or energetics.

Effect of isoflurane and propofol on myofibrillar Mg-ATPase activity. Isolated myofibrils from failing right ventricles were treated with either isoflurane (1.5%, n = 5) or propofol (50 μM, n = 5) for 20 minutes. Neither the maximal Ca2+-activated Mg-ATPase activity nor the Ca2+ required for half maximal Mg-ATPase activity was affected by the anesthetics.
Discussion
Isoflurane and propofol are widely used agents to induce or maintain anesthesia. Our study reveals that both isoflurane and propofol decrease force development in cardiac muscle isolated from rats with RV failure. Furthermore, we showed that the weakened force generation stems from a decrease in maximal Ca2+-activated force, especially at low-to-moderate doses. Decreased Ca2+ availability may also contribute to reducing force development, but only at high propofol concentrations. Finally, our data show that neither propofol nor isoflurane further reduces myofilament Mg-ATPase activity that is already below normal in the failing RV preparation.
Anesthetics and Myofilaments in the Failing Myocardium.
Myocardial contractility is jeopardized not only by Ca2+ dysregulation (Lehnart et al., 2009) but also by alterations in the myofilaments themselves (Kass and Solaro, 2006; van der Velden, 2011). For example, altered phosphorylation of troponin and other myofilament proteins can cause changes in myofilament Ca2+ responsiveness and thus alter force development in the failing heart/myocardium (Knott et al., 2002; van der Velden et al., 2003; Yuan et al., 2006; Bilchick et al., 2007). Oxidation of tropomyosin and actin are also important contributors to contractile dysfunction after ischemia and/or during overt HF (Canton et al., 2004, 2006; Duncan et al., 2005). As a result of these modifications, the failing myocardium often exhibits decreased myofilament Ca2+ responsiveness (i.e., decreased Fmax and increased Ca50) during steady-state activations (Stull et al., 2004; Dai et al., 2006; Belin et al., 2007; Tan et al., 2009).
Recently, we reported that both isoflurane and propofol decrease force development by directly targeting key residues in myosin heavy chain, actin, and myosin light chain (Meng et al., 2016). Consistent with what is observed in normal cardiac muscles, here we found that isoflurane and propofol also compromise force development in failing RV muscle preparations. Moreover, analysis of the steady-state force–Ca2+ relationship revealed some important mechanistic aspects regarding the means by which anesthetics affect force generation in the failing cardiac muscle. First and foremost, both anesthetics reduced Fmax but not Ca50. Second, the myofilament ATPase–pCa relationship was unaffected by these agents. Third, when delivered at high doses, propofol, but not isoflurane, reduced the [Ca2+]i transients. In aggregate, these observations support the notion that propofol and isoflurane alter myofilament properties in failing cardiac muscle preparations.
The fact that the anesthetics were able to depress force development further and reduce Fmax supports our recent finding that anesthetics exert their effects by binding directly to target myofilament proteins (Meng et al., 2016) rather than by acting through post-translational pathways, such as phosphorylation and oxidation. However, our current data show that these baseline changes in the myofilament properties of the failing myocardium affect the interaction of anesthetics with the myofilament proteins. For example, our evidence showing that the overall myofilament Mg-ATPase activity is not altered by anesthetics, unlike what occurs in normal muscle preparations, suggests that in a failing heart, anesthetics likely alter cross-bridge cycling but not the interaction between myosin and actin (i.e., cross-bridge formation). This possibility is in keeping with previous evidence showing that isoflurane does not affect myosin-driven actin mobility or maximum myofilament ATPase activity in isolated myosin/actin preparations (Vivien et al., 2005). Taken together, this evidence suggests that, in an HF preparation, anesthetics are likely to target additional myofilament proteins, such as the regulatory proteins troponin, tropomyosin, and myosin light chain. Therefore, our findings support the need for additional, more in-depth studies that evaluate how the interaction between these anesthetic agents and the myofilament proteins influences the cross-bridge cycling in the failing myocardium.
Anesthetics and [Ca2+]i Transient/Cycling in the Failing Myocardium.
A well known hallmark of HF is a decrease in the release of Ca2+ from the sarcoplasmic reticulum (SR) (Hasenfuss and Pieske, 2002; Piacentino et al., 2003). Factors that contribute to the defective SR function include dysfunction of SR Ca2+ ATPase and reduced SR Ca2+ ATPase levels (Hasenfuss, 1998; Hasenfuss et al., 1999; O’Rourke et al., 1999), phosphorylation of SR ryanodine receptor (Marks et al., 2002), and enhanced expression of the Na-Ca exchanger (Flesch et al., 1996; Ahmmed et al., 2000).
Here we observed that, at concentrations of 50 μM and above, propofol decreased intracellular transients, an effect that would account for the observed reduction in force development (Fig. 4). However, previous studies that have examined the impact of propofol on myocardial function have generated conflicting results. Early studies that used failing human trabecular muscles from atria and ventricles showed that propofol decreased force development and inhibited isolated SR Ca2+ uptake at high concentrations (≥56 μM) (Sprung et al., 2001). In the normal myocardium, propofol can increase Ca2+-activated ATPase activity, suggesting an increase in myofilament Ca2+ sensitivity (Kanaya et al., 2003). However, some of these previous investigations did not evaluate the possible influence of propofol on myofilament sensitivity. In fact, these studies were carried out without measuring force. Recently, we re-examined this issue by conducting experiments in which we determined force, sarcomere length, and intracellular cytosolic free Ca2+ simultaneously in intact cardiac trabecular muscles (Ding et al., 2011; Meng et al., 2016). We found that propofol lowered force development by decreasing myofilament Ca2+ responsiveness. In the present study, we found that high-dose propofol further decreased [Ca2+]i transients in failing RV muscle preparations, consistent with earlier findings obtained in failing human muscles, which showed that propofol (56 μM) decreased SR Ca2+ uptake (Sprung et al., 2001). Thus, propofol has dual adverse effects on cardiac excitation coupling in the failing myocardium: it decreases myofilament Ca2+ responsiveness at low-to-moderate concentrations and SR Ca2+ release at high concentrations.
Limitations and Perspective.
The previously described functional experiments were carried out at room temperature, not at body temperature. Measuring [Ca2+]i with our technique is extremely difficult at 37°C mainly because metabolism of fura-2 is extremely rapid at that temperature. Additionally, obtaining the steady-state force–[Ca2+]i relationship is virtually impossible at 37°C because SR cycling becomes too rapid. However, we have determined twitch force and corresponding [Ca2+]i at 37°C in the past and found similar effects by both agents on force and [Ca2+]i transients (Ding et al., 2011). Nevertheless, we would like to caution readers when drawing conclusions from results obtained at room temperature. Another caveat is that the anesthetic concentrations needed to observe the functional effects were slightly higher than those used clinically. For example, the blood concentration of propofol at which 50% of patients did not respond to skin incision was 18 μM in the presence of fentanyl (as opposed to 81 μM without fentanyl), and the concentration at which 95% of patients did not respond to skin incision was 28.9 μM in the presence of fentanyl as determined by high-performance liquid chromatography (Smith et al., 1994). For isoflurane, the blood concentration at which patients opened their eyes is 0.53% as determined by gas chromatography (Katoh et al., 1992). In the current paper, as well as in our prior studies (Ding et al., 2011; Meng et al., 2016), our effective propofol dose range was between 20 and 50 μM, and effective concentration of isoflurane was 1%–1.5%. We used 50 μM propofol and 1.5% of isoflurane for most of the experiments. However, this apparent discrepancy has technical and biologic explanations. First, the in vitro preparation is superfused rather than coronary perfused; second, the use of room temperature may reduce the efficacy of the drug; third, myocytes are, in general, more sensitive to any given drug at body temperature than they are at room temperature. Thus, future, in-depth studies are needed to evaluate the molecular mechanism that underlies the anesthetic-induced reduction of myofilament Ca2+ responsiveness in the failing myocardium. Recently, we carried out molecular docking studies based on proteomic data obtained in myofilaments treated with photo-labeled anesthetics and have identified several anesthetic binding sites in myosin and actin (Meng et al., 2016). Thus, based on acquired and current evidence, it is tempting to speculate that these anesthetic agents do not directly interfere with the modalities of interaction between myosin and actin. Instead, in HF cardiac muscle preparations, they are more likely to affect regulatory proteins such as troponins, tropomyosin, and myosin-binding protein C. Identifying the binding sites for anesthetics in the regulatory myofilament proteins is fertile terrain for future investigation. Future studies are also warranted to determine possible synergistic effects when the two anesthetic agents are used in combination.
Translationally speaking, the results from this rat pulmonary hypertension–induced (right) HF model will stimulate future in-depth clinical investigation into the effect of anesthetics on the dysfunctional right heart during pulmonary hypertension. Clinically, it has been recognized that anesthetic agents may further depress myocardial function, especially of the right heart, in patients with pulmonary hypertension (McGlothlin et al., 2012). However, this clinically relevant aspect needs to be investigated further, both in terms of the real magnitude of the anesthetic-imparted RV dysfunction and the effect on clinical outcome in patients with pulmonary hypertension/RV dysfunction requiring anesthesia.
Conclusion.
At low-to-moderate concentrations, anesthetics such as isoflurane and propofol reduce force development in failing cardiac muscle preparation by decreasing myofilament Ca2+ responsiveness. At high dosage, propofol, but not isoflurane, decreases force development by reducing intracellular Ca2+ availability. Our findings that anesthetics exert a negative inotropic effect teach us to exercise caution when delivering anesthesia to patients with HF or other cardiac diseases. Moreover, these data suggest that we make some selective choices among available anesthetics in patients with reduced cardiac performance. Ideally, these findings could create some interest for designing additional anesthetic agents devoid of this adverse effect on myofilament Ca2+ responsiveness and SR cycling, or in which these collateral effects are minimized.
Authorship Contributions
Participated in research design: Meng, Ren, Yu, Paolocci, Gao.
Conducted experiments: Meng, Ren, Chen, Gao.
Performed data analysis: Meng, Ren, Chen, Agrimi, Gao.
Wrote or contributed to the writing of the manuscript: Meng, Ren, Yu, Agrimi, Paolocci, Gao.
Footnotes
- Received May 3, 2019.
- Accepted September 11, 2019.
↵1 T.M. and X.R. contributed equally to this work.
↵This study was supported by grants from the Shangdong Provincial Natural Science Foundation [Grant ZR2017BH022] and China Postdoctoral Science Foundation [Grant 2019M652395] to T.M., a grant from the American Heart Association [Grant 17GRNT33670367] to W.D.G., and grants from the National Institutes of Health [Grant R01 HL136918 and R01 HL063030] to N.P.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- [Ca2+]i
- intracellular Ca2+
- Ca50
- [Ca2+]i required at 50% of Fmax
- FAC
- fractional area of change
- Fmax
- maximal Ca2+-activated force
- HF
- heart failure
- K-H
- Krebs-Henseleit
- MCT
- monocrotaline
- RV
- right ventricular
- SR
- sarcoplasmic reticulum
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}