


Visual Overview

Abstract
Endothelial dysfunction is a hallmark of diabetic vasculopathies. Although hyperglycemia is believed to be the culprit causing endothelial damage, the mechanism underlying early endothelial insult in prediabetes remains obscure. We used a nonobese high-calorie (HC)-fed rat model with hyperinsulinemia, hypercholesterolemia, and delayed development of hyperglycemia to unravel this mechanism. Compared with aortic rings from control rats, HC-fed rat rings displayed attenuated acetylcholine-mediated relaxation. While sensitive to nitric oxide synthase (NOS) inhibition, aortic relaxation in HC-rat tissues was not affected by blocking the inward-rectifier potassium (Kir) channels using BaCl2. Although Kir channel expression was reduced in HC-rat aorta, Kir expression, endothelium-dependent relaxation, and the BaCl2-sensitive component improved in HC rats treated with atorvastatin to reduce serum cholesterol. Remarkably, HC tissues demonstrated increased reactive species (ROS) in smooth muscle cells, which was reversed in rats receiving atorvastatin. In vitro ROS reduction, with superoxide dismutase, improved endothelium-dependent relaxation in HC-rat tissues. Significantly, connexin-43 expression increased in HC aortic tissues, possibly allowing ROS movement into the endothelium and reduction of eNOS activity. In this context, gap junction blockade with 18-β-glycyrrhetinic acid reduced vascular tone in HC rat tissues but not in controls. This reduction was sensitive to NOS inhibition and SOD treatment, possibly as an outcome of reduced ROS influence, and emerged in BaCl2-treated control tissues. In conclusion, our results suggest that early metabolic challenge leads to reduced Kir-mediated endothelium-dependent hyperpolarization, increased vascular ROS potentially impairing NO synthesis and highlight these channels as a possible target for early intervention with vascular dysfunction in metabolic disease.
SIGNIFICANCE STATEMENT The present study examines early endothelial dysfunction in metabolic disease. Our results suggest that reduced inward-rectifier potassium channel function underlies a defective endothelium-mediated relaxation possibly through alteration of nitric oxide synthase activity. This study provides a possible mechanism for the augmentation of relatively small changes in one endothelium-mediated relaxation pathway to affect overall endothelial response and highlights the potential role of inward-rectifier potassium channel function as a therapeutic target to treat vascular dysfunction early in the course of metabolic disease.
Introduction
Diabetes is a heterogeneous metabolic disease defined by constant hyperglycemia (American Diabetes Association, 2015). The most common forms of diabetes are type I diabetes, whereby autoimmune destruction of pancreatic beta cells leads to absolute insulin deficiency, and type II diabetes, where a combination of insulin deficiency and insulin resistance underpins hyperglycemia (American Diabetes Association, 2015). Type II diabetes is a growing epidemic, and its prevalence is expected to exceed 366 million people in 2030 (Wild et al., 2004). This alarming spread of diabetes is attributed to the accelerated shift toward Western diets rich in saturated fats and refined sugars (Kuhnlein and Receveur, 1996).
Atherosclerotic cardiovascular disease (ASCVD) is the primary cause of morbidity and mortality in type II diabetes (American Diabetes Association, 2017). Endothelial dysfunction (ED), which is widely known to be associated with vascular derangements, is an early independent predictor of ASCVD (Sitia et al., 2010; Mudau et al., 2012). The risk of ASCVD is not only restricted to patients with hyperglycemia, but also extends to prediabetic patients, who have worse cardiovascular outcomes compared with healthy individuals (Giráldez-García et al., 2015; Huang et al., 2016). Previous studies proposed various mechanisms that link ED to type II diabetes, such as impaired insulin signaling, oxidative stress, proinflammatory signaling, and the direct effect of hyperglycemia (Roberts and Porter, 2013). Research interest in identifying the onset of ED and the corresponding pathologic mechanisms early in the development of diabetes has grown significantly, particularly in the prediabetic stage. Deterioration of endothelial function starts at early stages of insulin resistance in the form of decreased NO bioavailability, impaired prostacyclin production, or increased endothelial vasoconstrictor production (Du et al., 2006; Picchi et al., 2006; Duncan et al., 2007; Polovina and Potpara, 2014). On the other hand, emerging evidence describes a role for endothelium-dependent hyperpolarization (EDH) in regulating the overall endothelial response, including NO-mediated relaxation (Fancher et al., 2018; Alaaeddine et al., 2019). While few studies recorded observational changes in EDH-type relaxation in animal models of insulin resistance and metabolic disease (Miller et al., 1998; Gradel et al., 2018), our knowledge still lacks a detailed mechanistic explanation behind the contribution of EDH to the integrative endothelial response in this stage. This is of particular importance since metabolic perturbations associated with diabetes have long been linked to increased L-type calcium channel activity and smooth muscle depolarization (Barbagallo et al., 1995; Ungvari et al., 1999; Nystoriak et al., 2014; Nieves-Cintrón et al., 2017). Thus, we hypothesize that relatively limited perturbations in EDH pathway occurring early in metabolic dysfunction could have a significant impact on endothelial function and myoendothelial feedback, via affecting other pathways in smooth muscle and endothelial cells including the NO pathway.
To test this hypothesis, we used a rat model of mild metabolic challenge developed in our laboratory (Al-Assi et al., 2018; Elkhatib et al., 2019). This rat model develops hyperinsulinemia and hypercholesterolemia in absence of obesity, hyperglycemia, and hypertension following 12 weeks of exposure to a high-calorie diet. However, a gradual increase in fasting and random blood glucose levels is observed after 16 weeks of feeding, indicating the eventual incidence of diabetic hyperglycemia. Vascular tissue from these rats demonstrated increased contractility and elevated production of reactive oxygen species. This model provides a reasonably wide window for studying functional changes occurring in the course of early metabolic alterations without interference from hyperglycemia, impaired glucose tolerance, or obesity. We were able to show a selective impairment in EDH-type relaxation in aortic vessels of these rats. This dysfunction appeared to be driven by a reduced inward rectifier potassium channel activity and involved increased production of reactive oxygen species, which in turn, potentially interfered with nitric oxide synthase function.
Materials and Methods
Ethical Approval.
All animal experiments were conducted according to an experimental protocol approved by the Institutional Animal Care and Use Committee at the American University of Beirut in compliance with the Guide for Care and Use of Laboratory Animals of the Institute for Laboratory Animal Research of the National Academy of Sciences.
Experimental Design.
Male Sprague-Dawley rats (5 to 6 weeks of age; 150 g) were randomly divided into three groups (seven rats per group): 1) rats fed with normal chow (control, 3 kcal/g), 2) rats fed with mild hypercaloric diet (HC, 4.035 kcal/g) for 12 weeks, and 3) rats fed with mild hypercaloric diet for 12 weeks and treated with 20 mg/kg atorvastatin once daily at week 8. All rats had free access of food and water throughout the 12-week period. Rats were kept in a temperature- and humidity-controlled room, in a 12-hour light/dark cycle. Body weight was measured weekly. The treatment group received atorvastatin incorporated with the HC chow. The calculated dose was geometrically mixed with a 5-g HC diet pellet. The drug-containing food pellet was administered once daily starting week 8 and continued for 4 weeks till the end of the 12-week feeding period. Ad libitum access to HC diet resumed after the drug-containing 5-g pellet was consumed. HC diet was administered to the HC-fed group in the same manner during that period.
Food Preparation and Macronutrient Composition.
HC diet was prepared as described previously (Al-Assi et al., 2018; Elkhatib et al., 2019). Normal chow diet (ENVIGO) was obtained from Teklad Rodent Diets (Madison, WI). This diet offers 3 kcal/g divided as follows: 54% from carbohydrates, 32% from protein, and 14% from fat (0.9% saturated fat by weight). The HC diet is prepared in-house and consists of food grade fructose (20% by weight; Santiveri Foods, Barcelona, Spain) and hydrogenated vegetable oil (Mazola, 15% by weight; BFSA, Yanbu, Saudi Arabia) added to the normal chow diet. Major electrolytes and vitamins were supplemented to match the concentration in ENVIGO diet and as recommended by the American Institute of Nutrition (Reeves et al., 1993). The final composition of the HC diet was confirmed by bomb calorimetry and found to be by weight (calorie content) 18.06% fat (38.68%, 5% saturated fat by weight), 15.8% protein (15.66%), and 46.13% carbohydrates (45.73%).
Blood Chemistry.
Total serum cholesterol, serum insulin, random blood glucose levels, and glucose tolerance were assessed in all groups. Rats were fasted in metabolic cages with free access to drinking water for either 6–8 hours prior to glucose tolerance testing or 12 hours for cholesterol measurement. Blood samples (0.7 ml) were taken by retro-orbital bleeding and centrifuged at 4000 rpm for 10 minutes. The supernatant serum was isolated and stored at −80°C till the time of analysis. Total serum cholesterol was measured using electrochemiluminescence on a Cobas 6000 reader (Roche Diagnostics, Basel, Switzerland). Measurement of rat insulin in serum was carried out using a rat insulin ELISA kit (Thermo-Fisher Scientific, Waltham, MA) according to manufacturer’s protocol. Intraperitoneal glucose tolerance was evaluated in fasting rats as described previously (Lozano et al., 2016) following a 2 g/kg intraperitoneal glucose load. Glucose was measured in blood droplets collected by a tail prick at baseline, 15, 30, and 60 minutes using Accu-check Performa glucometer (Roche Diagnostics, Rotkreuz, Switzerland).
Noninvasive Blood Pressure Measurement.
Noninvasive blood pressure measurement was done by tail-cuff using a CODA High Throughput Monitor (Kent Scientific, Torrington, CT) (Wang et al., 2017). Blood pressure data were collected at the end of week 11 of feeding. Any irregular or unacceptable recording noted as a false recording by the system was excluded. The parameters obtained are systolic, diastolic, mean arterial blood pressure, and heart rate.
In Vitro Aortic Vessel Reactivity.
At the end of the 12-week period, the rats were anesthetized by isoflurane and sacrificed by decapitation. The thoracic cavity was exposed and the thoracic aorta was isolated for tension measurements as outlined in previous studies (Plane et al., 2005; El-Mas et al., 2011; Kimmoun et al., 2015). Specifically, thoracic aorta was excised from beneath the aortic arch to immediately above the diaphragm and dissected free of connective tissue. Aortic rings 0.3–0.5 cm in length were used for contractility experiments. The rings were assigned to experiments in the same given order among all groups in such a way that rings from equivalent positions were used to perform the same assay across all groups. The rest of the rings were flash frozen in liquid nitrogen and stored at −80°C. Aortic rings were mounted in a multichannel organ bath system (World Precision Instrument, Inc., Sarasota, FL). Each ring was suspended in a 15-ml oxygenated Krebs solution organ bath (NaCl 118 mM, KCl 4.7 mM, MgSO4.7H2O 1.2 mM, NaHCO3 25 mM, CaCl2.2H2O 2.5 mM, KH2PO4 1.2 mM, glucose 11.1 mM; pH 7.4 upon aeration with 95%O2/5%CO2 gas mixture). The bath was constantly aerated with 95% O2/5% CO2 gas mixture and temperature was maintained at 37°C. Digital recording of changes in aortic tissue tension was performed using Transbridge TBM4M force transducer (World Precision Instrument, Inc.) and an Acqknowledge 3.9.1 data acquisition system (World Precision Instrument, Inc.). The aortic rings were exposed to a resting tension of 2 g and allowed to stabilize for 1 hour, during which rings were washed with Krebs solution and tension was adjusted.
Our recent findings in this rat model (Elkhatib et al., 2019) indicated signaling changes in the aortic smooth muscle layer in response to phenylephrine stimulation and increased ROS production, possibly interfering with endothelial function. Thus we opted to use a fixed concentration of the contractile agonist to expose the tissue to the same stimulus to be better able to assess the myoendothelial feedback. This approach was used previously despite changes in the initial contractile tone (Oyama et al., 1986; Pieper and Gross, 1988; Taylor et al., 1992; McNally et al., 1994; Keegan et al., 1995; Fleischhacker et al., 1999; Shaligram et al., 2018). Aortic tissues were constricted with phenylephrine (30 μM) or U-46619 (0.5 μM) (Plane and Garland, 1996). The concentration of PE used to constrict the aortic rings was selected within the upper submaximal range obtained from PE concentration-response curves conducted in preliminary experiments to accentuate the difference between control and HC-fed rats allowing dissection of the role of individual endothelial pathways. Nevertheless, to confirm the findings in experiments with different initial contractile tone, an additional set of experiments were conducted using equieffective concentrations of phenylephrine (10−6–10−5 M to produce ∼1 g tension) and a reduced concentration of U46619 (0.1 μM to produce ∼3 g tension). Aortic endothelial vasoreactivity was assessed by response to acetylcholine (ACh, 1 × 10−9–1 × 10−4 M) (Grizelj et al., 2015) or diazoxide (100 μM). Several concentrations of diazoxide, within the range of 1 × 10−7–1 × 10−4 M were tested and a concentration of 1 × 10−4 M was selected as it produced reproducible relaxations of a considerable magnitude. The sensitivity of ACh-mediated relaxation to a number of blockers (Table 1) was assessed. Effect of ACh was represented as percentage of residual constriction calculated based on 100% contraction of PE/U-46619.
Concentrations of different agents used in vascular reactivity experiments
Cerebral Arterial Pressure Myography.
After decapitation, the brain was immediately removed and placed in an ice-cold buffer solution containing (NaCl 130 mM, KCl 4 Mm, MgSO4.7H2O 1.2 mM, NaHCO3 4 mM, CaCl2.2H2O 1.8 mM, HEPES 10 mM, KH2PO4 1.18 mM, glucose 6 mM, EDTA 0.03 mM, pH 7.4). Rat middle cerebral artery was isolated from surrounding connective tissue and dissected into segments of 2 to 3 mm of length. Cerebral artery segments were mounted in a chamber attached to a pressure myograph (DMT, Hinnerup, Denmark) for measurement of arterial diameter. Arteries were allowed to warm up to 37°C and equilibrate for 15–20 minutes at 10 mmHg. The vessels were then pressurized to 60 mmHg and allowed to develop a stable myogenic tone over 30 minutes. Pressure was then dropped to 20 mmHg and then increased again to 80 mmHg in a series of pressure steps until a reproducible myogenic response is obtained. Endothelial function was evaluated using ACh (10 μM) in the presence or absence of potassium channel blocker BaCl2 (30 μM) at 80 mmHg. The vasodilatory effect of ACh was estimated as a percentage of the active tone produced at 80 mmHg defined as the difference in the vessel diameter in normal and calcium-free buffer solution with 2 mM EGTA.
Quantitative Polymerase Chain Reaction.
Quantitative polymerase chain reaction was performed as described previously (Al-Assi et al., 2018). Total RNA was extracted from aortic tissue using RNeasy Mini kit with DNase treatment (Qiagen, Hilden, Germany) and first-strand cDNA was produced with the Sensiscript RT kit (Qiagen) with oligo d(T) primer. Primer pairs for rat Kir2.1, Kir2.2, Cx37, Cx40, Cx43, and Cx45, and GAPDH were designed and obtained from Sigma (St. Louis, MO). Primer sequence was Kir2.1 (KCNJ2), forward AAAGCGTGTGTGTCTGAGGT, reverse ATCGGGCACTCGTCTGTAAC; Kir2.2 (KCNJ12), forward CCACTGACCGAGAAGTGCCC, reverse ATCGTAGCCCGTAGCCAATG; Cx37 (Gja4) forward TTGACCACCGAGGAGAGACT, reverse AGCCCCAGAGCCCTATACAT; Cx40 (Gja5) forward CCAGTCTCCAACACTTGGCA, reverse GCGGAAAATGAACAGGACGG; Cx43 (Gja1) forward TTCATTGGGGGAAAGGCGTG, reverse CTGGGCACCTCTCTTTCACTT; Cx45 (Gjc) forward GTTAACAGGGCAAACCAATTCCA, reverse AGATGGACTCTCCTCCTACCG and GAPDH forward AGACAGCCGCATCTTCTTGT and, reverse CTTGCCGTGGGTAGAGTCAT. Q-PCR was carried out with SYBR-Green and threshold cycle was established using Bio-Rad iCycler (Hercules, CA). Transcript abundance was computed by the 2−ΔΔCt method with GAPDH as a reference.
Western Blotting.
Western blotting was carried out as described in our previous studies (Moreno-Domínguez et al., 2014; El-Yazbi et al., 2015; Al-Assi et al., 2018). Briefly, aortic tissue samples stored at −80°C were crushed under liquid nitrogen. Fifty milligrams of aortic tissue was added to a 1 ml protein extraction buffer composed of 100 mM dithiothreitol, 1% SDS, 0.9% NaCl, and 80 mM Tris hydrochloride (pH 6.8). Preliminary experiments were done to optimize tissue amounts needed for protein extraction. Samples were heated at 95°C for 10 minutes and left overnight on a rocking shaker at 4°C. Aliquots of equal protein content were loaded on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and the separated proteins were transferred and fixed on a nitrocellulose membrane. Membranes were then blocked with 5% skimmed milk and TBST (Tris-buffered saline with 0.1% Tween 20) for 2 hours at room temperature (RT). The membranes were then incubated with a dilution of the primary antibody in 1% skim milk and 0.1% TBST (1:200 for rabbit polyclonal anti-Kir2.1, 1:500 for anti-phospho-Akt Thr308, and rabbit polyclonal anti-Akt; Abcam, Cambridge, UK; 1:1000 for rabbit polyclonal anti-Cx43, Sigma-Aldrich; 1:1000 for rabbit polyclonal anti-phospho-eNOS Ser1177 and rabbit polyclonal anti-eNOS; and 1:1000 for rabbit monoclonal anti-GAPDH; Cell Signaling, Danvers, MA) overnight at 4°C. Afterward, the membranes were washed with 0.02% TBST (4 × 5 minute) and incubated with 1:40,000 biotinylated goat anti-rabbit Ig secondary antibody (Abcam) in 0.1% TBST for 1 hour at RT. Membranes are then washed with 0.02% TBST (4 × 5 minutes) and incubated with 1:200,000 horseradish peroxidase-conjugated streptavidin (Abcam) in 0.1% TBST for 30 minutes at RT. After washing with 0.02% TBST (2 × 5 minutes) and TBS (2 × 5 minutes), membranes were developed using Clarity Western ECL substrate for 5 minutes prior to image detection by Chemidoc imaging system (BioRad). ImageJ software was used to measure optical density of protein bands. A ratio of arbitrary density units was obtained for the protein band of interest and the density of the band representing total protein for p-eNOS and p-AKt after stripping and reprobing, while eNOS, Cx43, and Kir2.1 bands were normalized to GAPDH. Membrane stripping was done using freshly prepared mild stripping buffer (15 g glycine, 1 g SDS, and 10 ml Tween in 1 liter of ultrapure water, pH 2.2). Membranes were inserted in enough volume of the buffer to be covered at RT for 5–10 minutes. Buffer was discarded and another volume was added for 5–10 minutes. Afterward, membranes were washed with 0.5% TBST (2 × 5 minutes) and then with TBS (2 × 5 minutes, and used for reprobing.
DHE Staining.
Aortic tissue sectioning and staining was done as described previously (Al-Assi et al., 2018). Dihydroethidium (DHE) staining was performed on cryosections to demonstrate reactive oxygen species (ROS) load. Fluorescent images were obtained using a Zeiss Axio inverted microscope (Carl Zeiss, Germany) through the Alexa Fluor 568 filter for the DHE red fluorescence and measured against the green collagen autofluorescence obtained through the Alexa Fluor 488 filter. Images were taken from three sections for each animal. The obtained images were analyzed using Zen software.
Chemicals.
All chemicals were obtained from Sigma unless otherwise indicated. Pharmaceutical grade atorvastatin was obtained as a kind gift from a regional pharmaceutical manufacturer (PharoPharma).
Statistics.
Data were expressed as mean ± S.E.M. Comparisons between groups were done using Student’s t test, one-way ANOVA followed by Tukey post hoc test, as well as two-way ANOVA followed by Sidak’s multiple comparisons test in comparing the effect of different concentrations among groups using GraphPad Prism software (San Diego, CA). The specific test used is mentioned in the corresponding figure legend. P value <0.05 was considered statistically significant.
Results
Metabolic Consequences of a 12-Week Mild Hypercaloric Feeding.
Full metabolic and hemodynamic profiling of this rat model was done previously (Al-Assi et al., 2018; Elkhatib et al., 2019). To confirm the metabolic characteristics in the cohort used in the present study, body weight, blood glucose, glucose tolerance, and serum insulin and cholesterol were assessed at the end of the 12-week feeding period. Similar to previous observations, no differences were detected in body weight (Fig. 1A), random blood glucose levels (Fig. 1B), glucose tolerance (Fig. 1C), and hemodynamic parameters including systolic blood pressure (Fig. 1F), diastolic blood pressure, and heart rate (data not shown) between control and HC-fed rats. However, as expected, HC-fed rats exhibited elevated plasma insulin (Fig. 1D) and total serum cholesterol levels (Fig. 1E) compared with the control group.

Metabolic and hemodynamic properties of HC-fed rats following 12-week mild hypercaloric feeding. (A) Body weight after 12 weeks of feeding compared with control; (B) random blood glucose level (BGL) on sacrifice day; (C) intraperitoneal glucose tolerance test; (D) fasting plasma insulin level; (E) fasting total serum cholesterol; and (F) systolic blood pressure measurement. Data presented are mean ± S.E.M. of seven separate observations. Statistical significance was assessed by unpaired Student’s t test for (A, B, and D–F) and by two-way ANOVA for (C). *P < 0.05 vs. the corresponding value in control rats.
Endothelium-Dependent Relaxation Is Impaired in HC-fed Rats with an Intact SNP-Mediated Relaxation.
To examine endothelium-dependent relaxation (EDR), in vitro contractility experiments were conducted on thoracic aortas obtained from control and HC-fed rats at the end of the 12-week feeding duration. Figure 2A depicts representative tracings of the relaxation of preconstricted aortic rings to increasing concentrations of ACh. A progressive relaxation was observed in rings from control rats that reached a maximum of ∼75% of the initial PE tone. On the other hand, aortic rings from HC-fed rats demonstrated an impaired ACh-mediated relaxation (∼25% of the PE tone), which leveled off at a lower ACh concentration (1 μM, Fig. 2B). Whereas the aortic rings from HC-fed rats exhibited an exaggerated contractile response upon exposure to PE (Fig. 2C, left), the maximal absolute tension reduction in response to ACh was less in rings from HC-fed rats confirming the endothelial deficit (Fig. 2C, right). Although previous studies suggested that initial contractile tone does not affect ACh-evoked EDR tone (Hansen and Nedergaard, 1999), we conducted an additional set of experiments with equieffective PE concentrations to rule out this possibility. In these experiments, there was a similar reduction in ACh-mediated relaxation in HC-fed rat aortic rings (Supplemental Fig. 1A). ACh failed to produce any relaxation in denuded rings of either group (data not shown), confirming the endothelial dependence of ACh-mediated relaxations. The NO donor SNP produced similar relaxation patterns in both groups (Fig. 2D). Furthermore, to investigate the possibility of release of the contractile endothelial mediator endothelin-1 as the underlying cause for the observed dysfunction in aortic rings from HC-fed rats, ACh-mediated relaxation was assessed in these rings in the presence of endothelin-1 receptor antagonist atrasentan (Fig. 2E). However, this intervention did not alter the aortic response to ACh observed in the HC group. Atrasentan was also without effect in preliminary experiments conducted on control tissue.

Impaired ACh-mediated endothelium dependent relaxation in HC-fed rats aortic segments. (A) Representative tracings of the relaxation response to increasing ACh concentrations. Dashed line represents the basal tension level before constriction with PE. Time points where ACh was added are marked by arrowheads; (B) summary of the ACh-evoked relaxation in aortic rings from seven control and seven HC rats; (C) absolute tension increase (left) and decrease (right) in response to PE and ACh, respectively, in experiments summarized in (B); (D) relaxant response to SNP in control (five rats) vs. HC-fed (five rats) aortic rings; (E) ACh-mediated relaxation in HC-fed rat aortic rings in presence (five rats) and absence (seven rats) of 10 nM atrasentan. Data presented are mean ± S.E.M. Statistical significance was assessed by unpaired Student’s t test for (C) and by two-way ANOVA for (A, B, D, and E). *P < 0.05 vs. the corresponding value in control rats in (B–D); and the corresponding values in absence of atrasentan in (E).
Impaired Endothelial Function in Aorta of HC-fed Rats Is Related to an Altered Endothelial Relaxing Mediator Profile.
We examined the relative contribution of different endothelial relaxing mediators in rat aorta and the differential impact of HC feeding on them. On one hand, blockade of endothelial NO production by l-NAME significantly reduced ACh-mediated relaxation in aortic rings from both rat groups (Fig. 3A). Yet a residual relaxation that gradually increased to ∼25% at higher ACh concentrations was observed only in aortic rings from control rats. No additional attenuation of the ACh-mediated relaxation was observed upon the combination of indomethacin and l-NAME (data not shown). To address the role of EDH-type relaxation, the effect of blockade of SK/IK with apamin and Tram-34 (Fig. 3B) and Kir channels with BaCl2 (Fig. 3B) was assessed. SK/IK channel blockers appeared to attenuate ACh-mediated relaxations in either group, albeit more effectively in aortic rings from control rats. Interestingly, while BaCl2 attenuated the ACh-mediated relaxation in rings from control rats, particularly toward the higher ACh concentrations, aortic rings obtained from HC-fed rats were not sensitive. Of note, the maximal residual ACh-mediated relaxation in tissues from control rats in presence of l-NAME was 24.56% ± 4.19%, which was not different from the maximal reduction of the ACh-mediated relaxation brought about by treatment with BaCl2 (33.37% ± 10.78%, P > 0.05, t test).

Effect of blockade of different endothelium-relaxing pathways on ACh-evoked relaxation in aortic rings from control (top panels) and HC-fed (bottom panels) rats. (A) ACh-evoked relaxation in presence (five rats) and absence (seven rats) of 100 μM l-NAME; (B) ACh-evoked relaxation in presence (five rats) and absence (seven rats) of 1 μM apamin and Tram-34; (C) ACh-evoked relaxation in presence (five rats) and absence (seven rats) of 30 μM BaCl2. Data presented are mean ± S.E.M. Statistical significance was assessed by two-way ANOVA. *P < 0.05 vs. the corresponding value in absence of the different blockers.
Importantly, incubation of aortic rings from control rats with l-NAME, apamin/Tram-34, or BaCl2 resulted in a significant increase in the PE-induced contraction. This was not observed in aortic rings from HC-fed rats, initially showing an increased PE-mediated constriction. As such, PE-induced tension was not different in treated rings from either control or HC-fed rats (control l-NAME: 3.91 ± 0.29 g, control apamin/Tram: 4.56 ± 0.95 g, control BaCl2: 3.98 ± 0.51 g, HC l-NAME: 4.32 ± 0.45 g, HC apamin/Tram: 3.17 ± 0.72 g, and HC BaCl2: 3.2 ± 0.23 g, P > 0.05, one-way ANOVA).
Impairment of Endothelial Function Is Likely Attributed to the Loss of Endothelium-dependent Hyperpolarization-type Relaxation.
To confirm the potential impairment of EDH-type relaxation, ACh-mediated relaxation profile of aortic rings was examined following pre-constriction with the thromboxane analog, U46619. This contractile agent produces vasoconstriction mainly through calcium sensitization (Plane and Garland, 1993), likely ruling out the potential vasodilatory effect of EDH. As predicted, aortic rings from both control and HC-fed rats produced equal contractions to U46619 and relaxed similarly to ACh (Fig. 4, A and B). Similar to prior studies (Kassan et al., 2013), the vascular contractile response to U46619 was much higher than that evoked by PE potentially masking the difference in ACh-evoked relaxation between control and HC-fed rat tissues. As such, an additional set of experiments was conducted using 0.1 μM U46619 to produce a contractile tension of ∼3 g (close to that produced by 30 μM PE). Nevertheless, these experiments showed no difference in ACh-mediated EDR between both groups (Supplemental Fig. 1B). Inhibition of NO production by L-NAME attenuated the relaxation response to ACh equally in aortic rings from both control and HC-fed rats (Fig. 4B), while BaCl2 was without effect on ACh-mediated relaxation (Fig. 4C).

ACh-evoked relaxation in aortic rings pre-constricted with the thromboxane analog, U46619. (A) Representative tracings of the relaxation response to increasing ACh concentrations (left). Time points where ACh was added are marked by arrowheads, and summary of the ACh-evoked relaxation in aortic rings from control and HC rats (right, five rats per group); (B) absolute tension increase (left) and decrease (right) in response to U46619 and ACh, respectively, in experiments summarized in (A); (C), ACh-evoked relaxation in presence and absence of 100 μM L-NAME in control (top) and HC-fed (bottom) rat aortic rings preconstricted with U46619 (five rats per group); (D) ACh-evoked relaxation in presence and absence of 30 μM BaCl2 in control (top) and HC-fed (bottom) rat aortic rings preconstricted with U46619 (five rats per group). Data presented are mean ± S.E.M. Statistical significance was assessed by unpaired Student’s t test for (B) and by two-way ANOVA for (A, C, and D). *P < 0.05 vs. the corresponding control value in (A and B) and the corresponding value in absence of the blockers in (C and D).
The Observed Endothelial Impairment in HC-fed Rats Is Likely due to Kir Channel Dysfunction.
Given the previous observations indicating the likelihood of Kir channel dysfunction in HC-fed rat aortic rings, we further examined their role as “end-stage amplifiers” of hyperpolarization (Sonkusare et al., 2016). We assessed the aortic response to the potassium channel opener diazoxide (Fig. 5A). Diazoxide produced an 80% tone reduction in control aortic rings preconstricted with PE; a response that was much attenuated in aortic rings from HC-fed rats. Interestingly, blockade of Kir channels with BaCl2 lacked any effect in HC-fed rat aortic rings, but brought the diazoxide-evoked relaxation in control rats to a level similar to that seen in the HC-fed group. Recent studies implicated Kir2.1 and 2.2 subunits as the molecular carrier of the inward rectifier potassium current in rat vasculature (Sancho et al., 2019). We examined the relative expression of both channel subunits in our vessel preparation using Q-PCR revealing that Kir2.1 was the predominant subunit expressed (Fig. 5B). While no difference in transcript level was observed among tissues from control and HC-fed rats (data not shown), Western blotting showed a reduction of Kir2.1 subunit expression in aortic tissues of HC-fed rats compared with control rats (Fig. 5C).

Dysfunctional Kir channels in HC-fed rat aortic rings. (A) Representative tracings for the diazoxide-mediated relaxations in control and HC-fed rat aortic rings (left). Arrowheads indicate the time point PE was added, while asterisks indicate the points where diazoxide was added; and average magnitude of these relaxations in presence and absence of 30 μM BaCl2 (right, rings from five rats in each experiment); (B) mRNA expression levels of Kir2.1 and Kir2.2 in control rat aortic tissue (data from four rats); (C) Kir2.1 protein expression level in aortic tissue of control vs. HC-fed rats (data from four rats). Data presented are mean ± S.E.M. Statistical significance was assessed by two-way ANOVA for (A) and unpaired Student’s t test for (B and C). *P < 0.05 vs. the corresponding control value in (A and C), and the Kir2.2 value in (B); #P < 0.05 vs. the value in rings unexposed to BaCl2.
Gap Junction Contribution to the Observed Endothelial Dysfunction in HC-fed Rat Aorta.
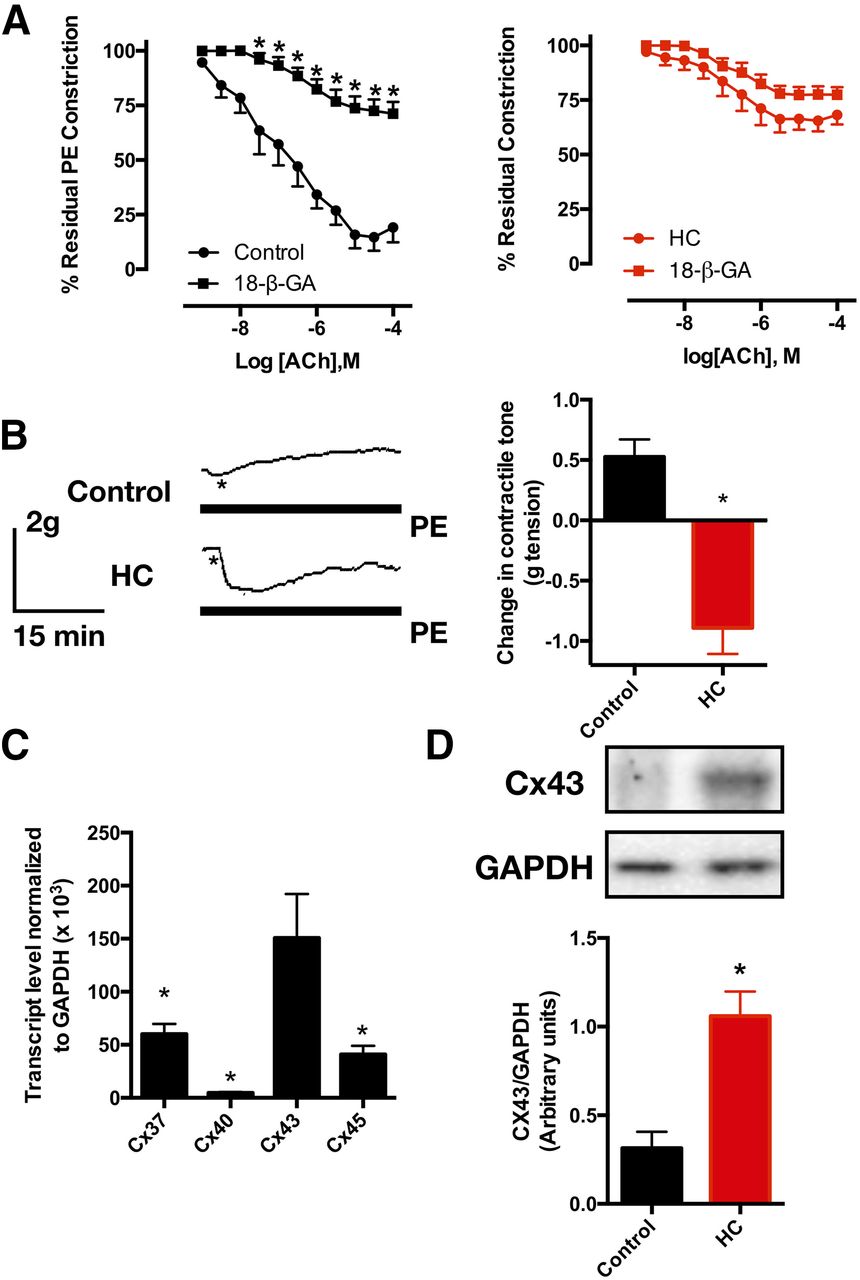
Since gap junctions are an essential element of myoendothelial feedback, we examined the effect of the gap junction blocker 18-β-GA on ACh-mediated relaxation (Fig. 6A). As expected, the gap junction blocker greatly attenuated the response to ACh in control aortic rings. Conversely, 18-β-GA did not alter the ACh-dependent relaxation in rings from HC-fed rats. Interestingly, the addition of 18-β-GA to the pre-constricted aortic rings produced disparate effects (Fig. 6B). Control rings exhibited an increase in contractile tone consistent with the interruption of EDH conduction and hence EDH-mediated relaxation. Surprisingly, rings from HC-fed rats demonstrated a decrease in vascular tone, indicating that gap junctions might mediate the conduction of an entity impairing endothelial function. On the other hand, the examination of the relative expression of connexin subtypes in our vessel preparation showed that Cx43 is the most abundant (Fig. 6C). As such, we proceeded to examine the relative expression of Cx43 between control and HC-fed rat aortic tissues. While there was no difference observed on the mRNA level (data not shown), Western blotting showed that Cx43 expression was higher in aortic tissues from HC-fed rats (Fig. 6D).
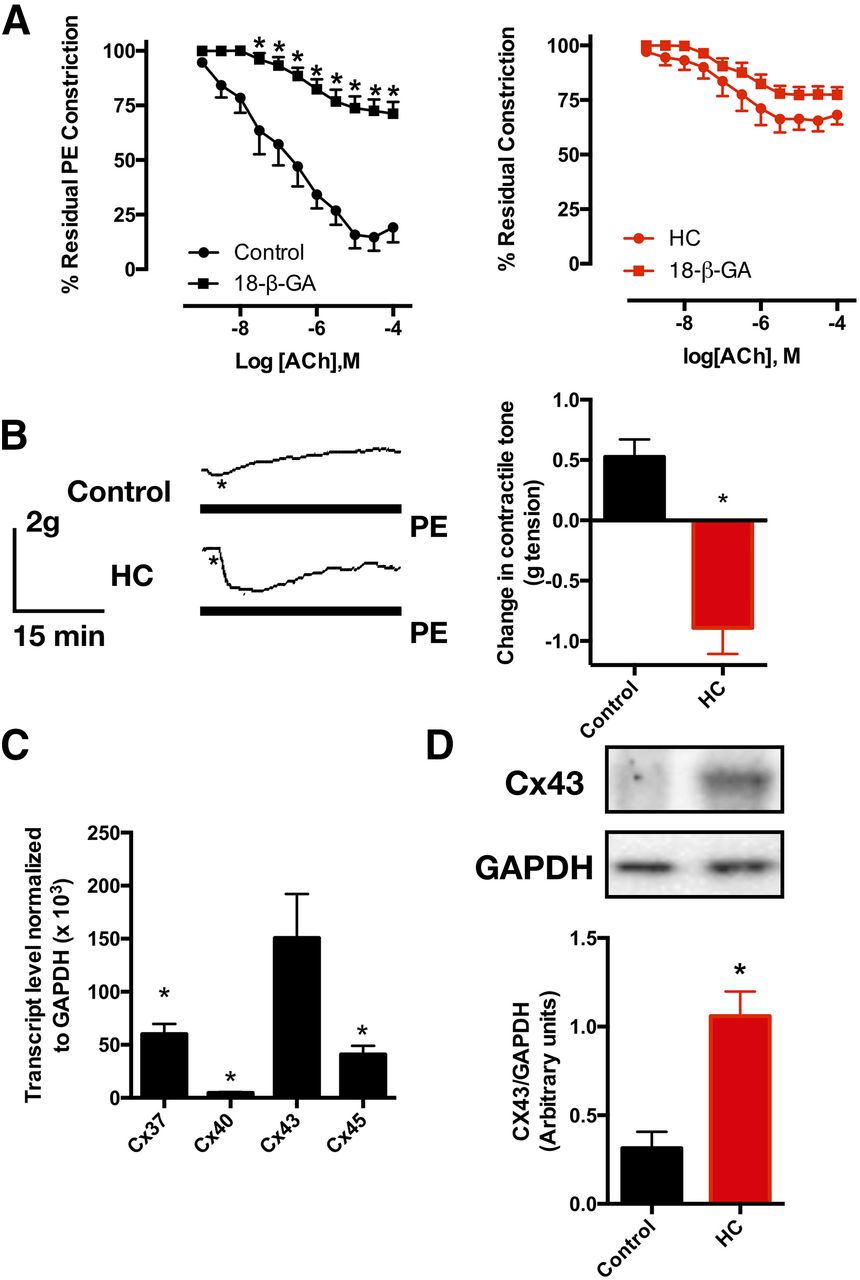
Effect of gap junction blockade on ACh-evoked relaxation and PE-induced constriction. (A) ACh-evoked relaxation in presence (five rats) and absence (seven rats) of 100 μM 18-β-GA in control (left) and HC-fed (right) rat aortic rings; (B) representative tracings of the effect of 100 μM 18-β-GA on PE-induced tone in control and HC-fed rat aortic rings (left). Asterisks represent the time points where 18-β-GA was added in the continued presence of PE, and average absolute tension change in these experiments (rings from five rats per group, right); (C) mRNA expression levels of Cx37, Cx40, Cx43, and Cx45 in control rat aortic tissue (data from four rats); (D) Cx43 protein expression level in aortic tissue of control vs. HC-fed rats (data from four rats). Data presented are mean ± S.E.M. Statistical significance was assessed by two-way ANOVA for (A) one-way ANOVA for (C) and unpaired Student’s t test for (B and D). *P < 0.05 vs. the corresponding value in absence of blocker in (A), control rat value in (B and D), and the Cx43 value in (C).
Reactive Oxygen Species Play a Role in the Impairment of Endothelium-dependent Relaxation in HC-fed Rats.
ROS examination using DHE staining showed a significant increase in aortic sections obtained from HC-fed rats (Fig. 7A). In vitro scavenging of ROS by incubating aortic rings from HC-fed rats with superoxide dismutase (SOD), not only reduced the ROS staining intensity, but also improved the ACh-mediated relaxation (Fig. 7B). SOD treatment was without effect on ACh-mediated relaxation in preliminary experiments done on control rat aortic rings. Since ROS was reported to spread across gap junctions among different vascular cell types (Feine et al., 2012) and are known to inhibit eNOS activity (Ohara et al., 1993), we investigated whether the reduction in tone observed post-gap junction blockade in HC-fed rat tissues was because of an interruption of eNOS inhibition by ROS. First, inhibition of eNOS in HC-fed rat aortic rings by preincubation with l-NAME precluded the loss of tone upon treatment with 18-β-GA (Fig. 7C). As well, ROS scavenging by SOD had a similar effect. To investigate whether the loss of Kir channel function was related to this observation, similar experiments were conducted in control rat aortic rings incubated with BaCl2 to block Kir channels. Not only did incubation with BaCl2 increase ROS upon DHE staining (Fig. 7D), BaCl2-treated control aortic rings also exhibited a loss of tone upon treatment with 18-β-GA (Fig. 7E) similar to the observations in HC-fed rat tissues. ROS staining persisted in HC-fed rat tissues or BaCl2-treated control tissues exposed to the gap junction blocker 18-β-GA in the same pattern observed in the above experiments (data not shown).

Aortic ROS levels and the effect of in vitro ROS modulation on endothelial function of rat aortic rings. (A) Representative micrographs of DHE staining of aortic tissues from control, HC-fed, and HC-fed rat aortic rings exposed to SOD in vitro (left), and average quantified data (right, data obtained from three sections per rat and four rats per group). DHE staining appears as red fluorescence on a background of green collagen autofluorescence. Scale bar, 40 μm; (B) representative tracings of the relaxation response to increasing ACh concentrations in aortic rings from HC-rats in presence (five rats) and absence (seven rats) of 200 U/ml SOD (left). Time points where ACh was added are indicated by arrowheads, and average responses at different concentrations (right); (C) representative tracings of the effect of 100 μM 18-β-GA on PE-induced tone in HC-fed rat aortic rings in the continued presence of PE, PE/l-NAME, or PE/SOD as indicated in the bar beneath the tracings (left). Asterisks represent the time points where 18-β-GA was added and average absolute tension change in these experiments (rings from five rats per group, right). (D) representative micrographs of DHE staining of aortic tissues from control rats with and without preincubation with BaCl2 (left), and average quantified data (right, data obtained from three sections per rat and four rats per group). Scale bar, 40 μm; (E) representative tracings of the effect of 100 μM 18-β-GA on PE-induced tone in control rat aortic rings in the continued presence of PE or PE/BaCl2 as indicated in the bar beneath the tracings (left). Asterisks represent the time points where 18-β-GA was added and average absolute tension change in these experiments (rings from five rats per group, right). Data presented are mean ± S.E.M. Statistical significance was assessed by one-way ANOVA for (A and C) two-way ANOVA for (B) and unpaired Student’s t test for (D and E). *P < 0.05 vs. the control rat value in (A) and the corresponding HC-fed rat values in (B and C).
Atorvastatin Treatment Improved Endothelium-dependent Hyperpolarization Profile.
Since hypercholesterolemia was shown previously to impair Kir channel function and reduce EDH (Fancher et al., 2018; Sancho et al., 2019), we examined the effect of blood cholesterol reduction in HC-fed rats on ACh-mediated relaxation in general and the BaCl2-sensitive component in particular. Four-week treatment of the HC-fed rats with the lipid-lowering agent atorvastatin restored their serum cholesterol levels to values comparable to that of the control group without affecting body weight, blood glucose, or serum insulin levels (Fig. 8A). This improvement was associated with an enhanced ACh-mediated relaxation compared with that of the HC-fed rat group (Fig. 8B). Significantly, BaCl2 attenuated the ACh-mediated relaxation in aortic rings from atorvastatin-treated HC-fed rats in a pattern similar to that observed in rings from control rats (Fig. 8C). Similarly, further indication of the restoration of Kir channel function was indicated by the increased diazoxide-mediated relaxation in atorvastatin-treated HC-fed rat aortic rings compared with their untreated counterparts (Fig. 8D). Alongside, Western blotting showed an increase of Kir2.1 subunit expression in the aorta of atorvastatin-treated rats (Fig. 8E). Moreover, DHE staining intensity indicative of ROS was also reduced by atorvastatin treatment (Fig. 8F).

Serum cholesterol lowering improves ACh-evoked relaxations and restores Kir channel function. (A) Effect of 4-week atorvastatin treatment in HC-fed rats on (left to right): total serum cholesterol, body weight, random blood glucose level, and plasma insulin level; (B) Representative tracings of the ACh-mediated relaxation in untreated (seven rats) and atorvastatin-treated (five rats) HC-fed rats (left). Arrowheads indicate time points where ACh was added and average responses and different concentrations (right); (C) effect of BaCl2 on ACh-mediated relaxation in atorvastatin-treated HC-fed rats (rings from five rats); (D) representative tracings for the diazoxide-induced relaxation of aortic rings from untreated and atorvastatin-treated HC-fed rats (left). Arrowheads indicate the time points where PE was added, while asterisks indicate the time points where diazoxide was added and average vasorelaxant response (five rats, right); (E) Kir2.1 protein expression in aortic tissues from untreated and atorvastatin-treated HC-fed rats (data for four rats); (F) representative micrographs of DHE staining of aortic tissues from untreated and atorvastatin-treated HC-fed rats and average quantified data. Data are obtained from three sections per rat and four rats per group. DHE staining appears as red fluorescence on a background of green collagen autofluorescence. Scale bar, 40 μm. Data presented are mean ± S.E.M. Statistical significance was assessed by one-way ANOVA for (A) two-way ANOVA for (B and C) and unpaired Student’s t test for (D–F). *P < 0.05 vs. the control rat value in (A) the corresponding value in untreated HC-fed rats in (B, E, and F) and the corresponding value in absence of blocker in (C).
Impairment of Endothelium-dependent Relaxation Is Not Related to Altered eNOS or Akt Expression/Phosphorylation.
To rule out the potential contribution of insulin resistance to the observed endothelial impairment, we investigated whether eNOS and Akt phosphorylation were altered in aortic tissues from HC-fed rats. Western blotting was used to examine the phosphorylated and total forms of either protein. Neither eNOS expression (Fig. 9A), phosphorylation (Fig. 9B), nor Akt phosphorylation (Fig. 9C) levels were altered in aortic rings from HC-fed rats.

HC-feeding does not alter Akt or eNOS phosphorylation. (A) Representative blots for total eNOS in aortic tissues from control and HC-fed rats together with average quantified data normalized to GAPDH (three rats per group); (B) representative blots for phospho-eNOS Ser1177 and total eNOS in aortic tissues from control and HC-fed rats together with average quantified data (three rats per group); (C) representative blots for phospho-Akt Thr308 and total eNOS in aortic tissues from control and HC-fed rats together with average quantified data (three rats per group). Data presented are mean ± S.E.M. Statistical significance was assessed by unpaired Student’s t test.
Endothelium-dependent Hyperpolarization Was Attenuated in the Microcirculation by HC Feeding and Restored by Atorvastatin.
Given the increased relative contribution of EDH in the microvasculature (Garland and Dora, 2017), we examined the endothelium-dependent relaxation pattern in middle cerebral arterioles from HC-fed rats to confirm our findings regarding EDH impairment. In pressure myography experiments, middle cerebral arteriole segments produced spontaneous myogenic tone in response to increased intravascular pressure. The vasodilatory response to 10 μM ACh was assessed across different groups. Cerebral arteriole segments from control rats showed considerable dilation in response to 10 μM ACh accounting for ∼40% of active tone generated by the vessel (Fig. 10A). Blocking Kir channels with 30 μM BaCl2 attenuated the ACh-induced relaxation to ∼10% of the active tone (Fig. 10D). Of note, addition of BaCl2 produced a phasic constriction indicating an interference with an ongoing hyperpolarization (Fig. 10, A and C). On the other hand, not only was ACh-mediated vasodilation much reduced in cerebral arteriole segments from HC-fed rats (∼10%, Fig. 10, A and B), but also this attenuated response was not affected by BaCl2 treatment (Fig. 10D). Moreover, the phasic contraction in response to Kir channel blockade by BaCl2 was blunted in vessel segments from HC-fed rats (Fig. 10C). Significantly, cerebral arterioles from atorvastatin-treated HC-fed rats demonstrated responses to ACh and BaCl2 that were similar to those from control rats.

Impaired ACh-evoked cerebral artery dilation in HC-fed rats is ameliorated by atorvastatin treatment. (A) Representative tracings of the cerebral arteriolar dilation to 10 μM ACh in presence and absence of 30 μM BaCl2 in vessel segments from control, untreated and atorvastatin-treated HC-fed rats. Time points where ACh was added are indicated by arrowheads, while time points were BaCl2 was added are indicated by asterisks. Tracings were recorded during continuous pressurization to 80 mmHg as indicated beneath them and Ca2+-free buffer solution was added as indicated by the bar toward the end of the recording; (B) average ACh-evoked relaxations (four rats/group); (C) average BaCl2-induced constrictions (four rats/group); (D) the effect of BaCl2 on ACh-mediated vasodilation (four rats/group). Data presented are mean ± S.E.M. Statistical significance was assessed by one-way ANOVA for (B and C) and two-way ANOVA for (D). *P < 0.05 vs. the control rat value in (B and C) and the value in absence of BaCl2 in (D).
Discussion
The optimal approach for mitigating diabetic vascular complications remains elusive (The Diabetes Control and Complications Trial Research Group et al., 1993; Heller, 2009; Genuth and Ismail-Beigi, 2012), especially since a significant proportion of diabetic patients present with established vascular complications at initial diagnosis (Fowler, 2008). As such, studying the early detrimental mechanisms contributing to ED in the course of diabetes development becomes paramount for effective management of diabetic vascular complications. Toward this end, we used a rat model of mild metabolic challenge, allowing a fairly wide window of opportunity to study vascular dysfunction prior to the development of hyperglycemia. To our knowledge, this is the first report to highlight an impairment of Kir channels as the primary cause of endothelial damage during early metabolic alterations, potentially triggered by hypercholesterolemia. Our results also highlight the potential interdependence among different endothelium-mediated vasodilatory pathways in producing integrated myoendothelial feedback, whereby a defect in one would be augmented due to a lack of positive interaction with the other.
The rat model used in the present study is a modified form of the high-fat diet-fed animal model, which permits the examination of the development of pathologic mechanisms in impaired glucose tolerance and early type II diabetes (Winzell and Ahrén, 2004). Our model receives a limited increase in daily calorie intake from fat. Data from previous studies in our laboratory showed that this diet produced stable fasting and random hyperglycemia after 16–20 weeks of treatment (Elkhatib et al., 2019). However, no changes in glucose tolerance, body weight, or blood pressure were observed at 12 weeks (Al-Assi et al., 2018; Elkhatib et al., 2019), thus permitting the study of functional changes occurring in the course of early metabolic alterations without interference from hyperglycemia, impaired glucose tolerance, obesity, or hypertension.
Whereas aortic rings from both control and HC-fed rats had similar vasodilatory responses to the exogenous NO donor sodium nitroprusside, HC-fed-rat aortic tissues showed an impaired ACh-mediated relaxation proposing that the observed defect was likely related to an endothelial, rather than a smooth muscle dysfunction. This was coupled with an increased initial contraction to 30 μM PE, indicating a potential dysfunction in myoendothelial feedback. The persistence of the same defective ACh-mediated relaxation pattern in HC-fed rat tissues in experiments where equieffective concentrations of PE were used rules out the possibility that the difference in preconstrictor tone affected the ACh response. While a rich body of literature describes a role for the increased production of endothelin-1, acting through ETA receptors, in ED observed in diabetes, obesity, and metabolic syndrome (Harris et al., 2008; Li et al., 2018; Schinzari et al., 2018; Samsamshariat et al., 2019), this did not appear to be the case in aortic rings from HC rats. In vitro exposure to the ETA receptor antagonist atrasentan did not alter aortic response to ACh. This was similar to previous observations in another animal model of early prediabetes where vascular response was not altered by endothelin-1 (Knudson et al., 2006). Indeed, the above studies citing a role for endothelin-1 in ED in diabetes or obesity reported these findings in advanced disease states with overt hyperglycemia and increased body weight, contrary to the observed phenotype in our rat model. In fact, even studies describing a role for endothelin-1 in impaired flow-mediated dilation in prediabetic patients linked the observed increase in plasma endothelin-1 to postprandial hyperglycemia (McDonald et al., 2019a,b), which was not observed in our model.
We then set to investigate the relative contribution of different EDR mechanisms and the effect of HC feeding on them. Similar to prior reports of endothelium-mediated vasodilation in conduit arteries (Joannides et al., 1995; Shimokawa and Godo, 2016), our results show that NO was the predominant mediator of EDR in aortic rings, but left a residual relaxation that corresponded to the contribution of EDH-type relaxation. EDH-type relaxation is typically described to be initiated by a calcium-triggered endothelial hyperpolarization mediated by SK and IK channel activation, which is followed by additional amplification of hyperpolarization through the action of Kir channels (Garland and Dora, 2017). In control aortic rings, the contribution of EDH-type relaxation to the ACh-induced EDR was confirmed in experiments where either SK/IK or Kir channels were blocked significantly attenuating ACh-mediated responses. Although unlikely based on the large difference between the apamin and Tram-34 concentration used and their functional IC50 values (Table 1), their lack of effect at the higher ACh concentrations could be due to a competitive relief of channel inhibition. The more pronounced effect of BaCl2 at higher ACh concentrations is in line with previous literature showing a more prominent role for Kir blockade in responses to higher ACh concentrations (McIntyre et al., 2001; Hangaard et al., 2015; Rasmussen et al., 2016).
On the other hand, while aortic rings from HC-fed rats appeared to have a relatively intact NO contribution to the reduced EDR, the lack of a residual relaxation, similar to that observed in the control rings, suggested a possible impairment of the EDH-type response. This was further confirmed in experiments using SK/IK and Kir channel blockers. Whereas apamin/Tram-34 reduced ACh response at low concentrations to some extent, albeit not as marked as in control tissues; BaCl2 did not alter the ACh effect implicating Kir channel dysfunction in the observed deficit. Significantly, PE-evoked contraction increased in control rings treated with apamin/Tram-34, BaCl2, or l-NAME. This was not the case in rings from HC-fed rats, suggesting that EDH impairment attenuated not only the feedback pathway mediated by Kir channels but also that driven by NO, in a first indication of the interplay between both pathways in our vessel preparation.
To further demonstrate that EDH impairment underlies the endothelial deficit in HC-fed rat aortic rings, we examined the ACh-mediated relaxation following preconstriction with the thromboxane mimetic U46619 (Plane and Garland, 1996). PE produces vascular contraction via calcium sensitization and membrane depolarization and increased cytosolic calcium levels (Plane and Garland, 1996). Under such circumstances, EDH would promote smooth muscle relaxation by opposing the agonist-induced depolarization. On the other hand, U46619-mediated contractions were accompanied by much less depolarization than that observed with PE, thus limiting contribution from EDH-type relaxation (Plane and Garland, 1996). As well, U46619 inhibits intracellular endothelial calcium increase (Ratnayake et al., 2018), potentially interfering with the activation of calcium-dependent potassium channels implicated as an early effector involved in EDH. Moreover, a previous study reported that thromboxane analogs reduced endothelial intercellular communication through gap junctions via increased Cx43 internalization (Ashton et al., 1999), additionally reducing the likelihood for conduction of EDH into the vascular smooth muscle (VSMC) layer. As such, not only was the ACh-mediated relaxation of the U46619 contractile tone insensitive to BaCl2, but also it was similar in aortic rings from both control and HC-fed rats. Therefore, these results confirm a selective impairment of EDH in aortic rings from HC-fed rats, which did not manifest once this relaxant mechanism was excluded. Of interest though, while initial constrictions to PE were higher in tissues from HC-fed rats, U46619 produced equal contractions in both groups. Indeed, our previous work showed that the nature of vasoconstrictor agent determined the functional components of myoendothelial feedback (Wei et al., 2018).
Kir channels are described as “end-stage amplifiers” of EDH (Sonkusare et al., 2016), owing to their essential role in amplifying hyperpolarization in VSMCs. The negative slope conductance property of the kir2.1 subunit allows Kir channels to respond to the increase in membrane hyperpolarization by enhancing its activity (Jantzi et al., 2006). Thus, we hypothesized that reduced EDH-type relaxation could potentially be attributed to defective Kir channel expression/function. As such, to emphasize the role of dysfunctional Kir channels, we adopted an approach previously used by Smith et al. (2008) and Allen et al. (2002), where a nonendothelium triggered hyperpolarization was induced by the application of the KATP channel opener diazoxide. Smith et al. (2008) showed that functional Kir channels were able to amplify this hyperpolarization and produce a sustained vasodilation, which was reduced upon pharmacological intervention with BaCl2 or if Kir 2.1 expression was low. This indeed was the case in our experiments where aortic rings from control rats responded by a more profound relaxation to diazoxide compared with rings from HC-fed rats, wherein the response to diazoxide was rendered insensitive to BaCl2 because of the potential deterioration of Kir channel function. Results from Western blots demonstrated a downregulation in protein content of Kir2.1 subunit in HC-fed rats, providing a molecular confirmation for the lack of Kir-mediated endothelial relaxing component.
An additional determinant of the vascular response to EDH-type relaxation is myoendothelial gap junctions that provide tight coupling among endothelial cells and VSMCs allowing for a bidirectional gate for electrotonic spread of charge and small molecules (Figueroa et al., 2004). Given the concluded reduction in myoendothelial feedback in HC-fed rat aortic tissue, suggested by the increased initial PE response and its lack of sensitivity to l-NAME and BaCl2, the effect of gap junction blockade by 18-β-GA on ACh-mediated relaxation was assessed. Congruent with a lack of EDH-type relaxation in HC-fed rat tissue, gap junction blockade did not affect the vasorelaxant response, which was the opposite of observations in control tissue where ACh responses were greatly attenuated. This was despite the observation that Cx43 subunit expression levels increased in aortic tissues from HC-fed rats. Cx43 upregulation is in line with previous literature showing that, while the vascular expression of some connexin subunits (Cx37 and 40) tended to decrease in metabolic disease, Cx43 expression was found to increase (Alaaeddine et al., 2019). Cx43 is highly expressed in VSMCs and endothelial cells, specifically at the myoendothelial junction (Abed et al., 2014) and is thought to increase in metabolic disturbance as a consequence of increased ERK phosphorylation (Ho et al., 2013), a common observation in the vascular tissue in our rat model (Al-Assi et al., 2018; Elkhatib et al., 2019). Paradoxically, while PE-constricted control tissues exposed to 18-β-GA responded by further constriction possibly due to the interruption of myoendothelial feedback, HC-fed rat aortic segments responded by tone reduction. This observation could be explained by previous evidence linking vascular depolarization to eNOS dysfunction. Previous studies showed that depolarization induced an increased NADPH oxidase-mediated production of ROS, leading to impairment of endothelial NO-mediated vasodilation (Sohn et al., 2000; Oelze et al., 2006). Cx43 was consistently implicated in the transfer of ROS across different cell types (Ramachandran et al., 2007; Hutnik et al., 2008; Taniguchi Ishikawa et al., 2012; Le et al., 2014; Raza et al., 2017) with protective or detrimental outcomes. The ability of ROS to spread among different vascular cell types across gap junctions was described (Feine et al., 2012) together with their well-documented role in inducing eNOS dysfunction (Ohara et al., 1993). Thus, it follows that smooth muscle depolarization, produced by PE, when left unopposed by EDH could potentially impair eNOS activity in a manner that is gap junction dependent. Under such circumstances, gap junction blockade will restore some eNOS activity by interrupting the detrimental outcome of myoendothelial communication. Indeed, this notion was supported by our current observation that tone reduction observed in HC-fed rat tissue following 18-β-GA exposure was abolished in tissues preincubated with l-NAME.
To further confirm and reconcile these findings, we assessed whether aortic tissues from HC fed rats showed an increased ROS production and if in vitro scavenging of ROS improved the relaxation response to ACh. Indeed, DHE staining showed an increased ROS level in the smooth muscle layer of HC-fed rat aortic tissues. In vitro exposure to the ROS scavenger SOD not only reduced ROS, but also improved ACh-evoked relaxation. This was also associated with a reversal of the tone reduction observed when gap junctions were blocked with 18-β-GA, since ROS no longer flowed into the endothelium and affected NOS function. Conversely, when reduced Kir channel function was simulated in control aortic rings treated with BaCl2, not only did ROS levels increase, but also the tone reduction associated with gap junction blockade was recapitulated, providing further support for the role of the interaction between vascular hyperpolarization, ROS production, and an integrated myoendothelial feedback. Interestingly, the persistence of DHE staining in the smooth muscle layer of HC tissues and BaCl2-treated control tissues after gap junction blockade possibly confirms that ROS production originated in smooth muscle.
To establish a link between these observations and changes occurring in vivo, we considered previous evidence describing a reciprocal relationship between Kir channel activity and levels of cholesterol in the microenvironment of the channel (Fang et al., 2006; Sancho and Welsh, 2018). Moreover, a recent study showed that hypercholesterolemia suppressed Kir2.1 channel function and attenuated flow-induced vasodilation (Fancher et al., 2018). Significantly, not only did cholesterol reduction by in vivo treatment with atorvastatin improve ACh-evoked relaxation in aortic tissues of HC-fed rats, the sensitivity of this relaxation to BaCl2 was also restored, indicating a possible amelioration of Kir channel dysfunction. Indeed, Western blotting showed an increase in the expression level of Kir2.1 channel. Along the same lines, vasorelaxation to diazoxide was also improved. It is noteworthy as well that ROS levels in the vascular smooth muscle layer were reduced in HC-fed rats treated with atorvastatin further confirming the proposed model.
Since our rat model is hyperinsulinemic, it could be argued that insulin resistance impairs the PI3K/Akt signaling pathway downstream of insulin receptor (Wheatcroft et al., 2003; Muniyappa and Sowers, 2013), possibly reducing Akt-mediated eNOS phosphorylation and eNOS activity leading to the observed endothelial phenotype. However, Western blotting showed no difference in the phosphorylation of Akt and eNOS in both groups. Moreover, the improvement in endothelial function observed in atorvastatin-treated HC-fed rats was not associated with an amelioration of hyperinsulinemia. Furthermore, examination of ACh-mediated effects in rat middle cerebral artery, as an example of a microvessel where EDH predominates the endothelial response (Garland and Dora, 2017), confirmed that the attenuated vasodilatory response in vessel segments from HC rats are rather a consequence of Kir channel dysfunction. Moreover, control vessels responded to BaCl2 exposure by a phasic constriction consistent with the blockade of functional Kir channels. This phasic contraction was greatly reduced in cerebral vessel segments from HC-fed rats. Significantly, atorvastatin treatment restored both the BaCl2-sensitive ACh-mediated vasodilation and the phasic contraction to BaCl2 indicating an amelioration of Kir channel function.
In conclusion, the present study demonstrates for the first time that early endothelial dysfunction in the course of metabolic disease is a consequence of impaired EDH-type relaxation due to a dysfunctional Kir2.1 channel activity. The endothelial deficit at this stage is further compounded by a potential reduction of eNOS activity, resulting from increased production of ROS in vascular smooth muscle accessing the endothelium through gap junctions. Correction of hypercholesterolemia restores Kir channel function, which opposes the smooth muscle layer depolarization, possibly reducing ROS production and improving overall endothelial response. Recognition of this early deficit highlights the role of Kir channel function as a potential therapeutic target for future drug development and emphasizes prophylactic statin treatment in improving vascular outcomes early in the course of metabolic disease.
Acknowledgments
The authors acknowledge the valuable help with experimental work offered by Lara Chaaban as a part of the Medical Research Volunteer Program of the American University of Beirut.
Authorship Contributions
Participated in research design: El-Yazbi, Plane.
Conducted experiments: Alaaeddine, Elkhatib, Mroueh.
Performed data analysis: Alaaeddine, Elkhatib, Mroueh, Fouad, Saad, El-Sabban, El-Yazbi.
Wrote or contributed to the writing of the manuscript: Alaaeddine, Elkhatib, El-Sabban, Plane, El-Yazbi.
Footnotes
- Received August 11, 2019.
- Accepted September 6, 2019.
↵1 R.A. and M.A.W.E. contributed equally to the study.
This study was supported by a Medical Practice Plan Research Grant offered by the Faculty of Medicine at the American University of Beirut to A.F.E. [Grant #320148]. R.A. is supported by a PhD Scholarship from the Faculty of Medicine at the American University of Beirut.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ACh
- acetylcholine
- ASCVD
- atherosclerotic cardiovascular disease
- Cx43
- connexin 43
- DHE
- dihydroethidium
- ED
- endothelial dysfunction
- EDH
- endothelium-dependent hyperpolarization
- EDR
- endothelium-dependent relaxation
- eNOS
- endothelial nitric oxide synthase
- HC
- high calorie
- 18-β-GA
- 18-β-glycyrrhetinic acid
- Kir
- inward-rectifier potassium channel
- l-NAME
- nitro-l-arginine-methyl ester
- NO
- nitric oxide
- PE
- phenylephrine
- ROS
- reactive oxygen species
- RT
- room temperature
- SK/IK
- small/intermediate calcium activated potassium channel
- SNP
- sodium nitroprusside
- SOD
- superoxide dismutase
- TBST
- Tris-buffered saline
- VSMC
- vascular smooth muscle cell
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}