


Abstract
The pregnane X receptor (PXR) is a ligand-activated nuclear receptor that acts as a xenobiotic sensor, responding to compounds of foreign origin, including pharmaceutical compounds, environmental contaminants, and natural products, to induce transcriptional events that regulate drug detoxification and efflux pathways. As such, the PXR is thought to play a key role in protecting the host from xenobiotic exposure. More recently, the PXR has been reported to regulate the expression of innate immune receptors in the intestine and modulate inflammasome activation in the vasculature. In the current study, we report that activation of the PXR in primed macrophages triggers caspase-1 activation and interleukin-1β release. Mechanistically, we show that this response is nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain-containing 3-dependent and is driven by the rapid efflux of ATP and P2X purinoceptor 7 activation following PXR stimulation, an event that involves pannexin-1 gating, and is sensitive to inhibition of Src-family kinases. Our findings identify a mechanism whereby the PXR drives innate immune signaling, providing a potential link between xenobiotic exposure and the induction of innate inflammatory responses.
Introduction
The pregnane X receptor (PXR) is a xenobiotic sensor that plays a key role in drug metabolism by regulating the expression of genes that encode enzymes responsible for drug detoxification and efflux (Koutsounas et al., 2013). As a member of the nuclear receptor (NR) superfamily, the PXR acts as a ligand-activated transcription factor, regulating gene expression in concert with its heterodimeric binding partner, the retinoid X receptor. In contrast to other NRs, the PXR’s ligand-binding domain exhibits a large flexible pocket that accommodates the binding of a variety of structurally unique ligands, including rifamycin antibiotics, pharmaceutical compounds, natural compounds, and contaminants of environmental origin (e.g., bisphenol A, organochloride pesticides) (Kliewer et al., 2002; Chang and Waxman, 2006; Staudinger et al., 2006; Gupta et al., 2008; Chang, 2009; Shukla et al., 2011).
The PXR is highly expressed in the liver and regions of the small and large intestine, and its role in regulating the host’s response to exogenous chemicals at these sites has been well characterized (Koutsounas et al., 2013) given their exposure to high concentration of exogenous ligands and xenobiotics. In addition, the PXR has been shown to regulate tissue inflammation through a reciprocal interaction with nuclear factor κ light chain enhancer of activated B cells (NF-κB) (Shah et al., 2007; Mencarelli et al., 2010, 2011). Indeed, we, and others, have reported that PXR agonists inhibit the release of inflammatory mediators from hepatocytes (Hu et al., 2010; Sun et al., 2015) and intestinal epithelial cells (Shah et al., 2007; Mencarelli et al., 2010, 2011) by inhibiting NF-κB–dependent signaling events, and can afford protection in experimental models of hepatic (Wang et al., 2010) and intestinal inflammation (Ma et al., 2007; Shah et al., 2007; Dou et al., 2012, 2013; Terc et al., 2014; Garg et al., 2016). The PXR can also regulate the expression/function of innate immune pattern recognition receptors within the intestinal epithelium, an effect that contributes to the proper maintenance of intestinal mucosal homeostasis and barrier function (Venkatesh et al., 2014).
Beyond the gastrointestinal tract, the PXR may contribute to the regulation of inflammation in other cell types (Schote et al., 2007; Zhou et al., 2009; Dubrac et al., 2010; Casey and Blumberg, 2012; Beyer et al., 2013). In the context of innate immunity, in contrast to its reported anti-inflammatory effects, Wang et al. (2014a) found that stimulation of the PXR in cultured vascular endothelial cells enhanced the expression of a number of innate immune receptors, including Toll-like receptors 2, 4, and 9, as well as nucleotide-binding oligomerization domain (NOD)-like receptor family members nucleotide-binding oligomerization domain, leucine-rich repeat, and CARD domain-containing 1, and nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain-containing 3 (NLRP3). Furthermore, endothelial cells treated with PXR agonists displayed features of NLRP3 inflammasome activation (Wang et al., 2014a).
The NLRP3 inflammasome is an innate immune signaling complex that mediates the responses to a variety of pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) (Mangan et al., 2018). The NLRP3 inflammasome’s activation following PAMP exposure and the induction of the ensuing innate immune response play key roles in host defense against viral, bacterial, and fungal pathogens (Mangan et al., 2018), whereas the recognition of DAMPs and subsequent activation of the NLRP3 inflammasome play an important role in the initiation of sterile inflammatory responses following tissue damage. Interestingly, several studies have reported the independent contributions of the PXR (Wang et al., 2014b) and NLRP3 inflammasome activation (Imaeda et al., 2009) in the pathogenesis of acute liver injury caused by sterile inflammation. In this scenario, the interplay between direct hepatocyte damage, DAMP release, and activation of resident macrophages is thought to contribute to the sterile inflammatory response that propagates acute liver damage (Hoque et al., 2012). Although the response of the hepatocyte has been the focus of much research, the role of PXR signaling and NLRP3 inflammasome within the macrophage, as a driver of inflammation, has not been addressed.
In the current study, we sought to test the hypothesis that the PXR plays a critical role in the macrophage by modulating NLRP3 inflammasome activation. In this study, we report that exposing primed human or mouse macrophages to their respective PXR agonists triggers caspase-1 activation and interleukin (IL)-1β secretion through a NLRP3-dependent mechanism. PXR-induced NLRP3 inflammasome activation was abolished by apyrase and selective inhibition of the P2X purinoceptor 7 (P2X7 receptor). Lastly, PXR ligands triggered a rapid and significant release of ATP, an effect that is dependent on pannexin-1 and Src kinase activation.
Materials and Methods
Reagents
PXR Agonists.
For experiments in mouse macrophages, the rodent selective PXR agonist pregnenolone 16α-carbonitrile (PCN; Sigma-Aldrich Canada, Oakville, Ontario, Canada) was prepared, as described above. For experiments in human macrophages, the human-selective PXR agonists rifaximin and SR12813 (Sigma-Aldrich Canada) were dissolved in sterile dimethylsulfoxide and added to culture media to attain the final experimental concentrations. As a vehicle control, identical volumes of dimethylsulfoxide were added and did not exceed a concentration of 1% v/v in media. The addition of exogenous ATP (5 mM; Sigma-Aldrich Canada) was used as a positive control in all experiments assessing inflammasome activation, as we have done previously (Ng et al., 2010; Hirota et al., 2011).
Inhibitors.
To assess the mechanism(s) involved in PXR agonist-induced NLRP3 inflammasome activation, we used the following compounds: to target reactive oxygen species (ROS), we employed the broad-spectrum antioxidant diphenyleneiodonium chloride (DPI; 100 μM) (Jabaut et al., 2013) (Sigma-Aldrich Canada); to chelate intracellular Ca2+ and target its effect in NLRP3 inflammasome activation, cells were pretreated with 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) (10 μM) (Ainscough et al., 2015) (Sigma-Aldrich Canada); and to assess the role of extracellular ATP and P2X7 receptor activation, we used apyrase (to break down extracellular ATP; 30 U/ml) (Iyer et al., 2009) (Sigma-Aldrich Canada) or treated cells with oxidized ATP (to selectively inhibit the P2X7 receptor oxidized ATP (oATP; 100 μM) (Grahames et al., 1999) (Sigma-Aldrich Canada). To assess the role of pannexin-1 in PXR-related ATP release, we used the panx-1 mimetic inhibitory peptide 10Panx (400 μM) (Grahames et al., 1999) (Tocris/Cedarlane, Burlington, Ontario, Canada) along with a scrambled peptide control ScPanx (400 μM) (Grahames et al., 1999) (Tocris/Cedarlane). To assess the role of Src-family kinases (SFKs) in the induction of ATP release, we used 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) (10 μM) (Chang et al., 2012) (Sigma-Aldrich Canada).
Cell Culture
Human Studies.
The human acute monocytic leukemia cell line (THP-1; American Type Culture Collection/Cedarlane, Burlington, Ontario, Canada) was propagated in RPMI 1640 medium supplemented with 10% fetal bovine serum and 50 μM 2-ME. To differentiate THP-1 cells into macrophages for inflammasome activation experiments, cells were plated at 5 × 105 cells/well in a 24-well plate in complete RPMI media and differentiated with phorbol-12-myristate-13-acetate (PMA; 50 ng/ml; Invivogen/Cedarlane, Burlington, Ontario, Canada) for 24 hours, as we have done previously (Ng et al., 2010; Hirota et al., 2011). NLRP3−/− THP-1 cells were generated and propagated, as described previously (Lau et al., 2018).
Mouse Studies.
Peritoneal macrophages were isolated from Nlrp3−/− and PXR−/− mice and their littermate wild-type counterparts (male, 8–10 weeks of age; all bred in house) 48 hours after receiving an i.p. injection of 4% thioglycolate-injected mice (BD Biosciences, San Jose, CA), as we have done previously (Ng et al., 2010; Hirota et al., 2011). Isolated macrophages were plated in complete RPMI media at 5 × 105 cells/well of a 24-well plate overnight and stimulated with 100 ng/ml ultra-pure lipopolysaccharide (LPS; Invivogen) in serum-free Opti-MEM for 30 minutes before challenge. For experiments with knockout mice, littermates were used as the wild-type control group for all experiments to control for potential microbiota-dependent differences in phenotype. All studies were approved by the University of Calgary’s Health Sciences Animal Care Committee (protocol AC15-0181). All approved activities conform to the guidelines and regulation for laboratory animal use set forth by the Canadian Council for Animal Care.
Assessing Inflammasome Activation
Western Blots.
Prior to performing inflammation activation experiments, isolated macrophages were pulsed with ultra-pure LPS (100 ng/ml for 30 minutes; Invivogen/Cedarlane). PMA-differentiated or LPS-pulsed mouse peritoneal macrophages were treated with their respective PXR agonists. Following the designated treatment period, culture supernatants were collected, cells were washed with ice-cold phosphate-buffered saline, and cell lysates were isolated following incubating the cells with lysis buffer (150 mM NaCl, 20 mM Tris, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, and protease inhibitor cocktail; phosphatase inhibitor cocktail, Complete Minitab; Complete PhoStop, Roche/Sigma-Aldrich Canada). Total protein was quantified using the Precision Red Advanced Protein Assay (Cytoskeleton/Cedarlane, Burlington, Ontario, Canada), and sample protein concentration was equalized. Culture supernatant and cell lysate samples were resolved, transferred to polyvinylidene difluoride membranes (0.2-μm pores; Bio-Rad Laboratories, Mississauga, Ontario, Canada), and blotted with the following antibodies: anti–caspase-1 (sc-622, sc-56036, and sc392736; Santa Cruz Biotechnology, Dallas, TX). Densitometry was performed using ImageJ, and cleaved caspase-1 was expressed as a percentage of pro–caspase-1.
IL-1β Secretion.
To quantify IL-1β release from PMA-differentiated or LPS-pulsed mouse peritoneal macrophages treated with their respective PXR agonists, culture supernatants were subject to enzyme-linked immunosorbent assay (human, DY201; mouse, DY401; R&D Systems/Cedarlane, Burlington, Ontario, Canada).
Assessing ATP Release
To characterize the mechanism by which PXR agonist triggered inflammasome activation, in some experiments, PMA-differentiated or LPS-pulsed mouse peritoneal macrophages were treated with their respective PXR agonists, culture supernatants were collected, and ATP was quantified using CellTiter-Glo Luminescent Cell Viability Assay (Promega North America, Madison, WI), as per the manufacturer’s instructions and described previously (Mortimer et al., 2015).
Statistical Analysis
All data were assessed for distribution using D’Agostino–Pearson normality test prior to statistical analysis using GraphPad Prism. Multiple comparisons of parametric data were accomplished using an analysis of variance, followed by Tukey’s post hoc test. For nonparametric data, or experiments with small samples sizes (N < 5), a Kruskal–Wallis test was used, followed by a Mann–Whitney test with a Bonferroni correction for multiple comparisons. In all experiments, N denotes individual experiments performed in different cell passages or cells derived from unique animals.
Results
PXR Agonists Trigger Caspase-1 Activation and IL-1β Secretion from Macrophages in a NLRP3-Dependent Manner.
Recent reports suggest that the PXR can regulate NLRP3 inflammasome activity in cultured vascular endothelial cells (Wang et al., 2014a, 2017), but this has not been assessed in macrophages, the prototypical model for innate immune signaling. To test the hypothesis that stimulation of the PXR triggers NLRP3 inflammasome activation in primed macrophages, we first treated LPS-primed peritoneal macrophages or PMA-differentiated THP-1 cells with their respective species-specific PXR ligands at concentrations previously reported to elicit selective responses in other cell types (Garg et al., 2016). In primed mouse macrophages, treatment with the rodent-specific PXR agonist PCN (for 6 hours) was able to trigger the release of IL-1β (Fig. 1A). In human macrophages, stimulation with two structurally unique selective human PXR agonists (SR12813 or rifaximin; for 6 hours) also triggered significant IL-1β secretion (Fig. 1, B and C).

Mouse and human PXR agonists trigger IL-1β release from primed macrophages. (A) The rodent-specific PXR agonist PCN (10 and 100 μM for 6 hours) elicits significant IL-1β release from LPS-primed peritoneal mouse macrophages, as measured by enzyme-linked immunosorbent assay. Human-specific PXR agonists (B) SR121813 (SR 0.1, 1, 4, 10 μM; 6 hours) or (C) rifaximin (rifx 1, 5, 10, 100 μM; 6 hours) stimulated significant IL-1β release from wild-type PMA-differentiated THP-1 cells. N = 3 to 4; *P < 0.05 for indicated comparison generated by analysis of variance and Tukey’s post hoc test.
To test whether IL-1β secretion in response to PXR activation in mouse and human macrophages was triggered by NLRP3 inflammasome activation, we next examined the ability of PXR agonists to trigger caspase-1 activation and IL-1β secretion in cells lacking NLRP3. When treated with PCN (1, 10, and 100 μM; for 6 hours), LPS-primed peritoneal macrophages isolated from Nlrp3−/− mice failed to trigger caspase-1 activation and IL-1β secretion (Fig. 2, A–C). The human-selective PXR agonists (SR12813, 4 μM; rifaximin, 5 and 10 μM; for 6 hours) also failed to activate caspase-1 and the subsequent secretion of IL-1β in PMA-differentiated THP-1 cells lacking NLRP3 (Fig. 2, D–F).
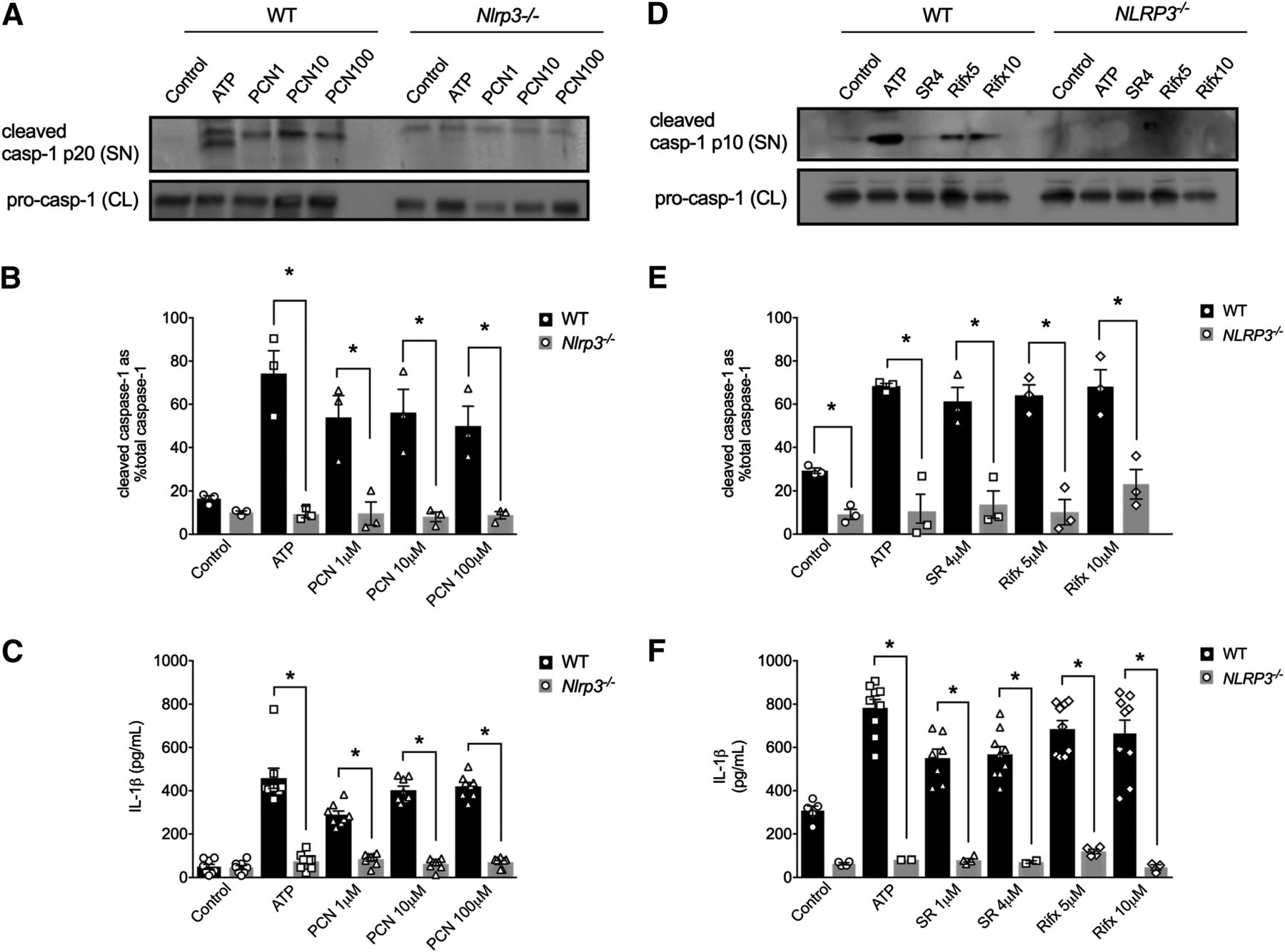
Mouse and human PXR agonists evoke caspase-1 activation and IL-1β release in a NLRP3-dependent manner. ATP (5 mM; positive control) and PCN (1, 10, and 100 μM) trigger caspase-1 activation [as assessed by detecting the cleaved caspase-1 p20 subunit in the supernatant (A and B)], along with significant IL-1β release from wild-type (WT) peritoneal macrophages (C), effects that were absent in macrophages isolated from Nlrp3−/− mice (A–C). ATP (5 mM; positive control), SR121813 (SR, 4 μM), and rifaximin (Rifx, 5 and 10 μM) trigger caspase-1 activation [as assessed by detecting the cleaved caspase-1 p10 subunit in the supernatant (D and E)], along with significant IL-1β release (F) from wild-type PMA-differentiated THP-1 cells, effects that were absent in NLRP3-deficient THP-1 cells (D–F). CL, cell lysate; SN, supernatant. All outcomes were measured after a 6-hour treatment period. N = 3–6; *P < 0.05 for indicated comparison generated by analysis of variance and Tukey’s post hoc test; Western blots are representative of three separate experiments.
Because antagonists of the human and mouse PXR are lacking specificity and exhibit off-target effects, we used macrophage cells isolated from PXR−/− mice (Fig. 3A) to validate the role of this receptor in the observed NLRP3 inflammasome responses. In support of our hypothesis, LPS-primed peritoneal macrophages isolated from PXR−/− mice did not exhibit caspase-1 activation nor secrete IL-1β in response to the selective mouse PXR agonist PCN (10 and 100 μM; Fig. 3, B–D), highlighting the role of the PXR in the activation of the inflammasome within macrophages.

Mouse PXR agonists fail to evoke caspase-1 activation or IL-1β release in peritoneal macrophages isolated from PXR−/− mice. The PXR is expressed in LPS-pulsed peritoneal macrophages (A) and is required for PCN-induced caspase-1 activation [as assessed by detecting the cleaved caspase-1 p20 subunit in the supernatant (B and C)] and subsequent IL-1β release (D), measured by enzyme-linked immunosorbent assay. CL, cell lysate; SN, supernatant. All outcomes were measured after a 6-hour treatment period. N = 3–6; *, denotes P < 0.05 for indicated comparison generated by analysis of variance and Tukey’s post hoc test; Western blots are representative of three separate experiments.
PXR-Mediated NLRP3 Inflammasome Activation Does Not Involve ROS or Intracellular Ca2+.
There are a variety of mechanisms that contribute to NLRP3 inflammasome activation, including, but not limited to, the generation of ROS, mobilization of intracellular Ca2+, and ATP-dependent K+ efflux via activation of the P2X7 receptor. In a human colon cancer cell line, the expression of the PXR enhanced sensitivity to oxidative stress that was associated with increased agonist-induced ROS production (Gong et al., 2006). To assess the role of ROS in the activation of the NLRP3 inflammasome by the PXR in macrophages, we used the broad-spectrum antioxidant DPI treatment (100 μM) (Jabaut et al., 2013). In PMA-differentiated THP-1 cells, DPI pretreatment had no effect on SR12813- or rifaximin-induced IL-1β secretion (Supplemental Fig. 1A), suggesting the ROS production does not mediate the PXR-induced inflammasome activation within macrophages.
Activation of the vitamin D receptor (VDR), a nuclear receptor closely related to the PXR, has been linked with increasing intracellular Ca2+ concentrations via an inositol 1,4,5-trisphosphate-dependent process (Tien et al., 1993) in epithelial cells, as well as activation of the NLRP3 inflammasome in macrophages (Tulk et al., 2015). Furthermore, Ainscough et al. (2015) reported that intracellular Ca2+ and its interaction with calmodulin were required for nigericin-induced IL-1β secretion in macrophages. To test whether intracellular Ca2+ plays a role in NLRP3 inflammasome activation mediated by the PXR, macrophages were pretreated with BAPTA-AM (10 μM), a cell membrane–permeable Ca2+ chelating agent. As reported previously, BAPTA-AM significantly reduced ATP-induced IL-1β secretion (Brough et al., 2003), but had no effect on the activation of the inflammasome by PXR agonists SR12813 and rifaximin (Supplemental Fig. 1B), suggesting mobilization of intracellular Ca2+ is not playing a role in PXR-mediated NLRP3 inflammasome activation.
Activation of the PXR Triggers ATP Release, Which Mediates NLRP3 Inflammasome Activation.
Reports have characterized the activation of the NLRP3 inflammasome through the autocrine/paracrine signaling of ATP released into the extracellular environment via pannexin-1 (Hung et al., 2013; Mortimer et al., 2015). Although there is no reported role for PXR inducing the release of ATP, bile acids, agents known to activate a variety of nuclear receptors, including the PXR, have been shown to induce ATP release in liver cells (Nathanson et al., 2001). Furthermore, activation of the farnesoid X receptor (FXR), a nuclear receptor closely related to the PXR, by bile acids induces rapid release of ATP in pancreatic cell lines (Kowal et al., 2015). Taken together, we next sought to test the hypothesis that PXR-mediated NLRP3 inflammasome activation could be driven by the release of ATP and subsequent activation of P2X7. First, to identify a role for ATP in our system, we cotreated PMA-differentiated THP-1 cells with apyrase (30 U/ml) to break down extracellular ATP (Iyer et al., 2009). As in our previous experiments, SR12813 and rifaximin triggered IL-1β secretion, but this response was abolished in macrophages cotreated with apyrase (Fig. 4A). As extracellular ATP is known to activate the NLRP3 inflammasome by triggering the K+ efflux through the P2X7 receptor (Hung et al., 2013; Mortimer et al., 2015), we next treated macrophages with oATP (100 μM), a selective P2X7 receptor antagonist (Grahames et al., 1999). As with apyrase cotreatment, oATP significantly attenuated IL-1β secretion in response to PXR agonists (Fig. 4B). Importantly, neither apyrase nor oATP had any effect on cell viability over the course of our experiments (Supplemental Fig. 2). Taken together, these data suggest that the PXR-mediated activation of the NLRP3 inflammasome involves the release of ATP and its subsequent activation of the P2X7 receptor.

PXR-associated NLRP3 inflammasome activation is sensitive to ATP degradation and inhibition of the P2X7 receptor. (A) Apyrase (APY; 30 U/ml) pretreatment, to degrade extracellular ATP, significantly reduces SR12813 (SR, 4 μM)- and rifaximin (Rifx, 10 μM)-induced IL-1β release from PMA-differentiated THP-1 cells. (B) oATP (100 μM), a selective P2X7 receptor antagonist, significantly reduces SR12813 (SR, 4 μM)- and rifaximin (Rifx, 10 μM)-induced IL-1β release from PMA-differentiated THP-1 cells. All outcomes were measured after a 6-hour treatment period. N = 6; *P < 0.05 for indicated comparison generated by analysis of variance and Tukey’s post hoc test.
PXR Stimulation Triggers the Rapid Release of ATP from Macrophages via Pannexin-1.
To further strengthen the link between the PXR, ATP release, and NLRP3 inflammasome activation, we directly quantified ATP release from mouse peritoneal macrophages (wild-type versus PXR−/−) and PMA-differentiated THP-1 cells at different time points. In macrophages isolated from wild-type mice, the rodent-specific PXR agonist PCN triggered rapid and significant extracellular ATP release that could be detected within 15 seconds and was sustained for up to 60 seconds before tapering off after 300 seconds (5 minutes). ATP release following PCN treatment was completely absent in cells isolated from PXR−/− mice (Fig. 5A). Similar ATP release responses were observed in THP-1 cells treated with either SR12813 or rifaximin (Fig. 5B). Collectively, these data indicate that the activation of the PXR triggers a rapid release of ATP that fits the kinetics reported by other NLRP3 inflammasome activators (Mortimer et al., 2015).

PXR agonists trigger rapid and significant ATP release from mouse and human macrophages. (A) PCN, a mouse PXR agonist, triggers rapid and significant ATP release from LPS-pulsed peritoneal macrophages isolated from wild-type mice, an effect that is absent in cells isolated from PXR−/− mice. N = 3; *P < 0.05 for wild-type (WT) PCN-treated macrophages compared with naive and PXR−/− cells generated by analysis of variance and Tukey’s post hoc test. (B) Human PXR agonists, rifaximin (Rifx) and SR12813 (SR), trigger rapid and significant ATP release from PMA-differentiated THP-1 cells. N = 3; *P < 0.05 for indicated comparison generated by analysis of variance and Tukey’s post hoc test.
The transmembrane channel pannexin-1 has been implicated in ATP release and NLRP3 inflammasome activation in macrophages (Hung et al., 2013; Mortimer et al., 2015), and thus we sought to determine whether this mechanism was at play following PXR stimulation in our studies. First, to assess the role of pannexin-1 in PXR-driven ATP release, PMA-differentiated THP-1 cells were exposed to pannexin-1–blocking peptide (10Panx; 400 μM) (Grahames et al., 1999) or a scrambled peptide control (ScPanx; 400 μM) prior to stimulation with SR12813 or rifaximin. PXR activation triggered rapid and significant ATP release in the scrambled peptide-treated cells (Fig. 6A), a response that was significantly attenuated by pannexin-1 channel blockade with 10Panx (Fig. 6A), without affecting cell viability (Supplemental Fig. 2).

Blockade of pannexin-1 and inhibition of SFKs attenuates human PXR agonist-induced ATP and IL-1β release from PMA-differentiated THP-1 cells. (A) Selective inhibition of pannexin-1 with a pannexin-1 channel-blocking peptide (10Panx, 400 μM; or scrambled control peptide ScPanx) abolishes rifaximin (Rifx)- and SR12813 (SR)-induced ATP release from PMA-differentiated THP-1 cells. (B) Inhibition of SFKs with PP2 (10 μM) significantly reduces rifaximin (Rifx)- and SR12813- (SR)-induced ATP release from PMA-differentiated THP-1 cells. N = 4; *P < 0.05 for indicated comparison generated by analysis of variance and Tukey’s post hoc test.
Although nongenomic roles for other NRs have been described, little is known about how the PXR regulates intracellular signaling processes within the cytosol. Others have reported the involvement of SFKs in the cytosolic effects of NRs (Vertino et al., 2005; Buitrago and Boland, 2010; Peng et al., 2012; Buitrago et al., 2013). Interestingly, receptor-mediated gating of pannexin-1 has been shown to involve SFK-dependent phosphorylation events (Lohman et al., 2015; Weilinger et al., 2016). To assess the role of SFKs in PXR-driven ATP release and NLRP3 inflammasome activation, we pretreated PMA-differentiated THP-1 cells with the Src-kinase inhibitor PP2 (10 μM) (Chang et al., 2012) and exposed them to PXR agonists. Selective inhibition of SFKs abolished the rapid ATP release triggered by SR12813 and rifaximin (Fig. 6B). Taken together these data suggest that PXR agonists trigger ATP efflux through a SFK- and pannexin-1–dependent process.
Discussion
In the current study, we found that stimulation of the PXR in primed macrophages triggered the activation of the NLRP3 inflammasome, resulting in caspase-1 activation and IL-1β secretion. Mechanistically, PXR activation–induced NLRP3 inflammasome signaling was reliant on pannexin-1–dependent ATP release and subsequent stimulation of the P2X7 receptor, a well-characterized driver of inflammasome activation (Mangan et al., 2018). In this process, PXR-dependent ATP release occurs as early as 15 seconds following receptor stimulation and involves SFK signaling, suggesting a cytosolic function for the PXR in macrophages. Altogether our data support a novel role for the PXR in triggering a host–defense response in macrophages, linking xenobiotic sensing and innate immunity in a system that may protect against xenobiotics and other chemical contaminants of foreign origin.
The PXR is a member of the nuclear receptor superfamily, which includes such members as the FXR, VDR, glucocorticoid receptor, and retinoid X receptor (Germain et al., 2006). The PXR, which is highly expressed in the liver and in intestinal epithelial cells, is best characterized for its ability to regulate the expression of enzymes involved in drug metabolism, detoxification, and excretion (Koutsounas et al., 2013). The PXR is also expressed in a variety of immune cells, including T cells, macrophages, and dendritic cells (Schote et al., 2007), and its signaling has been reported to modulate their function through mechanisms that are less understood (Zhou et al., 2009; Dubrac et al., 2010; Casey and Blumberg, 2012; Beyer et al., 2013). More recently, the PXR’s direct and indirect regulation of innate immune signaling has been reported in a number of systems. In a seminal report, Venkatesh et al. (2014) found that the PXR functions as a negative regulatory of Toll-like receptor gene expression in the intestinal epithelium, thereby indirectly modulating innate immune signaling in the intestinal mucosa. These data build on a body of literature that suggests that PXR can negatively regulate NF-κB–dependent inflammatory signaling in a variety of cell types (Xie and Tian, 2006; Zhou et al., 2006; Moreau et al., 2008).
In contrast to the notion that the PXR exhibits solely anti-inflammatory effects, Wang et al. (2014a) reported that the PXR could enhance NLRP3 inflammasome activity in vascular endothelial cells. The authors found that PXR stimulation upregulated the expression of NLRP3 and pro–IL-1β, and that prolonged stimulation triggered caspase-1 activation and IL-1β processing indicative of NLRP3 inflammasome activation. Although the kinetics of activation reported in this study do not conform to the traditional view of the NLRP3 inflammasome as an expeditious effector, our data suggest that PXR activation can trigger immediate ATP efflux from macrophages, a prerequisite for inflammasome activation in response to a variety of stimuli (Mangan et al., 2018). Indeed, the notion that a nuclear receptor can activate intracellular signaling events that culminate in rapid cellular responses is not without precedent. For instance, activation of the FXR by bile acids induces rapid release of ATP in pancreatic cell lines (Kowal et al., 2015). Tulk et al. (2015) also reported that stimulation of the VDR in primed macrophages triggered rapid NLRP3 inflammasome activation and IL-1β release. Our data suggest PXR stimulation involves the rapid gating of pannexin-1 through a SFK-dependent mechanism, resulting in ATP efflux and subsequent P2X7 receptor activation to trigger NLRP3 inflammasome activation.
Although our findings support a role for the PXR in initiating cell signaling that activates the NLRP3 inflammasome to induce IL-1β processing and release, others have reported contrasting observations. Sun et al. (2015) reported that pretreating hepatocytes with PXR agonists attenuated LPS-induced IL-1β release. Furthermore, a recent report by Wang et al. (2017) described a model wherein statins inhibit inflammasome activity in vascular endothelial cells, through the PXR-dependent inhibition of NF-κB–driven NLRP3 gene transcription. Some clarity could be provided to these disparities by interpreting them in context of the temporal nature of inflammasome activation. NLRP3 inflammasome output (i.e., IL-1β processing and release) requires two signals. The first stimulus, often termed signal 1, often involves Toll-like receptor–driven NF-κB activation to prime the cells, inducing the expression of the inflammasome components, including NLRP3 and pro–IL-1β (Mangan et al., 2018). In order for a functional NLRP3 inflammasome to elaborate, a second signal (also termed signal 2) is required, which usually takes the form of a DAMP or PAMP, and results in NLRP3 oligomerization, capsase-1 activation, resulting in IL-1β processing, and release (Mangan et al., 2018). The wealth of data implicating the PXR as a negative regulator of NF-κB signaling suggests its inhibitory effect on IL-1β release may be due to its inhibition of signal 1 (i.e., inhibiting the induction of NLRP3 and pro–IL-1β expression). Indeed, Luo et al. (2017) reported that pretreating macrophages with baicalein, an agent reported to activate the PXR (Dou et al., 2012), attenuated LPS-induced NLRP3 and pro–IL-1β expression, through its inhibition of NF-κB signaling. In this context, baicalein’s inhibition of signal 1 attenuated subsequent ATP-induced IL-1β secretion (Luo et al., 2017). It is important to highlight that in our studies we assessed the impact of PXR activation in primed macrophages that have already received signal 1. Thus, in the context of a primed cell that exhibits abundant expression of NLRP3 and pro–IL-1β, PXR stimulation acts as signal 2, gating pannexin-1 to allow the efflux of ATP, which culminates in caspase-1 activation and IL-1β release (Fig. 7).

Proposed model for the events that link PXR activation to NLRP3 inflammasome activation and IL-1β release. Ligation of the PXR (and heterodimerization with the retinoid X receptor) triggers the release of intracellular ATP through pannexin-1 channels. Extracellular ATP then binds to the P2X7 receptor, an event that prompts the assembly of the NLRP3 inflammasome and subsequent activation of caspase-1. Caspase-1 then cleaves pro–IL-1β into its active, secreted IL-1β.
Although the functional impact of the PXR’s modulation of NLRP3 inflammasome activation has yet to be elucidated, xenobiotic- and endobiotic-sensing mechanisms are thought to add an additional level of defense in the gastrointestinal tract of multicellular organisms (Dussault and Forman, 2002). For example, Caenorhabditis elegans upregulates xenobiotic response genes and initiates avoidance behaviors in the presence of pathogens and/or specific pathogenic factors in a process believed to enhance survival in the context of infection or exposure to environmental contaminants (Melo and Ruvkun, 2012; Jones et al., 2013). Mechanistically, these responses are mediated by a family of nuclear receptors that exhibit functional similarities to the mammalian PXR (Jones et al., 2013). As a ligand-activated receptor, the PXR’s flexible binding domain allows a variety of receptor–ligand interactions to occur (Kliewer et al., 2002; Chang and Waxman, 2006; Staudinger et al., 2006; Gupta et al., 2008; Chang, 2009; Shukla et al., 2011). Thus, the interplay between xenobiotics and innate immune signaling through the PXR may be broad reaching. Furthermore, the impact of endogenous PXR ligands of microbial origin and the regulation of intestinal mucosal inflammasome signaling require further attention. Ultimately, our findings suggest the existence of a complex interplay between xenobiotic-sensing mechanisms and innate immunity that may function as a conserved mechanism to protect the host from exposure to chemical agents of foreign origin. Additional work will be required to determine the functional role for this interplay in the context of health and disease.
Acknowledgments
We thank J. Staudinger (University of Kansas, Lawrence, KS) for providing Pxr−/− (Nr1i2−/−) mice.
Notes
Authorship Contributions
Participated in research design: Hirota, Mani, Chang, Hudson.
Conducted experiments: Hudson, Flannigan, Venu, Alston, Sandall.
Contributed new reagents or analytic tools: Sandall, MacDonald, Muruve.
Performed data analysis: Hudson, Hirota, Flannigan.
Wrote or contributed to the writing of the manuscript: Hirota, Hudson, Flannigan, Chang, Mani, MacDonald.
Footnotes
- Received December 5, 2018.
- Accepted April 8, 2019.
S.A.H.’s salary is supported by the Canadian Institutes of Health Research’s (CIHR) Canada Research Chair (CRC) program [Tier II CRC in Host–Microbe Interactions and Chronic Disease]. S.A.H.’s laboratory is supported by an infrastructure grant provided by the Canadian Foundation for Innovation John R. Evans Leaders Fund; operating funds from Crohn’s & Colitis Canada (S.A.H. and T.K.H.C. coinvestigators); the Dr. Lloyd Sutherland Investigatorship in IBD/GI Research; and Natural Sciences and Engineering Research Council (NSERC) Discovery Grant [RGPIN-2016-03842]. S.M.’s laboratory is supported by National Institutes of Health [Grants CA161879 and CA222469], U.S. Department of Defense [W81XWH-17-1-0479], and Broad Medical Research Program–Crohn’s & Colitis Foundation Investigator Award [Proposal 262520]. J.A.M. and D.A.M. are supported by the CIHR Health Challenges in Chronic Disease Signature Initiative [THC-13523]. C.F.S. holds scholarship awards from the Natural Sciences and Engineering Research Council and the Cumming School of Medicine. K.L.F. received support from the Beverly Philips postdoctoral fellowship through the Snyder Institute for Chronic Diseases at the University of Calgary and is currently supported by postdoctoral fellowships from Alberta Innovates and the Canadian Association of Gastroenterology/CIHR/Abbvie. T.K.H.C.’s laboratory is supported by a NSERC Discovery Grant [RPGIN-2014-03734].
The authors declare no competing or commercial interests.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- BAPTA-AM
- 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- DAMP
- danger-associated molecular pattern
- DPI
- diphenyleneiodonium chloride
- FXR
- farnesoid X receptor
- IL
- interleukin
- LPS
- lipopolysaccharide
- NF-κB
- nuclear factor κ light chain enhancer of activated B cells
- NLRP3
- nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain-containing 3
- NOD
- nucleotide-binding oligomerization domain
- NR
- nuclear receptor
- oATP
- oxidized ATP
- PAMP
- pathogen-associated molecular pattern
- PCN
- pregnenolone 16α-carbonitrile
- PMA
- phorbol 12-myristate 13-acetate
- PP2
- 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- PXR
- pregnane X receptor
- P2X7
- P2X purinoceptor 7
- ROS
- reactive oxygen species
- SFK
- Src-family kinase
- VDR
- vitamin D receptor
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}