


Abstract
Thiazolidinediones (TZDs) are peroxisome proliferator-activated receptor γ (PPARγ) agonists that represent an effective class of insulin-sensitizing agents; however, clinical use is associated with weight gain and peripheral edema. To elucidate the role of PPARγ expression in endothelial cells (ECs) in these side effects, EC-targeted PPARγ knockout (PpargΔEC) mice were placed on a high-fat diet to promote PPARγ agonist–induced plasma volume expansion, and then treated with the TZD rosiglitazone. Compared with Pparg-floxed wild-type control (Ppargf/f) mice, PpargΔEC treated with rosiglitazone are resistant to an increase in extracellular fluid, water content in epididymal and inguinal white adipose tissue, and plasma volume expansion. Interestingly, histologic assessment confirmed significant rosiglitazone-mediated capillary dilation within white adipose tissue of Ppargf/f mice, but not PpargΔEC mice. Analysis of ECs isolated from untreated mice in both strains suggested the involvement of changes in endothelial junction formation. Specifically, compared with cells from Ppargf/f mice, PpargΔEC cells had a 15-fold increase in focal adhesion kinase, critically important in EC focal adhesions, and >3-fold significant increase in vascular endothelial cadherin, the main component of focal adhesions. Together, these results indicate that rosiglitazone has direct effects on the endothelium via PPARγ activation and point toward a critical role for PPARγ in ECs during rosiglitazone-mediated plasma volume expansion.
Introduction
Thiazolidinediones (TZDs), including marketed rosiglitazone (Avandia), are synthetic insulin-sensitizing drugs that mediate their antidiabetic effects through peroxisome proliferator-activated receptor γ (PPARγ). TZDs also lower blood pressure and improve endothelial function (Parulkar et al., 2001; Haffner et al., 2002). Despite these beneficial effects, clinical use of TZDs (particularly in Europe) is limited by several adverse effects associated with their use (Gale, 2001). For instance, TZDs increase body weight gain in humans (2 to 3 kg for every percentage decrease in HbA1c values), an effect mainly attributed to increased subcutaneous fat depot size (Yki-Järvinen, 2004). Since TZDs exert their insulin-sensitizing effects via PPARγ activation, and given the well established role of this receptor in promoting adipogenesis (Tontonoz et al., 1994; Spiegelman et al., 1997), such an effect is likely mechanism-based (Kliewer et al., 1992, 2001; Keller et al., 1993; Wahli et al., 1995; Berger and Moller, 2002; Wang et al., 2004; Ricote and Glass, 2007; Campbell et al., 2008).
TZDs also promote fluid retention or edema, which may partially contribute to increases in total body weight (Yki-Järvinen, 2004). The incidence of edema is higher in patients treated with a combination of TZDs and insulin (Delea et al., 2003). In some cases, mild fluid retention can be treated by reducing the TZD dose and/or adding a diuretic (Hollenberg, 2003), although the majority of patients are not responsive to diuretics (Chen et al., 2005). TZD-induced edema may be receptor-mediated since structurally distinct, non-TZD PPARγ agonists also cause plasma volume expansion (PVE) in rodents (Berger et al., 2003). Likewise, the PPARγ Pro12Ala variant is a risk factor for PPARγ dual agonist–induced edema in type 2 diabetes patients (Hansen et al., 2006). Importantly, body weight gain and edema are associated with increased risk for congestive heart failure (2.5 times greater) in patients receiving combination therapy that includes a TZD and insulin (Delea et al., 2003). However, the Food and Drug Administration (FDA) lifted their restrictions on rosiglitazone after re-evaluation of the initial clinical trial data and continuous monitoring did not show heart infarct risks associated with the drug (Home et al., 2009; http://www.fda.gov/Drugs/DrugSafety/ucm376389.htm; http://www.fda.gov/Drugs/DrugSafety/ucm476466.htm). Given the pleiotropic beneficial effects of TZDs, it is crucial that the underlying mechanisms contributing to edema are elucidated.
Two independent groups showed that targeted disruption of PPARγ in collecting ducts confers resistance to TZD-induced fluid retention and PVE in mice (Guan et al., 2005; Zhang et al., 2005). Transcriptional regulation by PPARγ of Scnn1g, the gene encoding the epithelial Na(+) channel, reportedly plays a role in TZD-induced fluid retention by regulating renal salt absorption (Pavlov et al., 2009; Bełtowski et al., 2013). Another group reported that increased TZD-induced fluid retention is independent of the epithelial Na(+) channel (Vallon et al., 2009). Beyond fluid retention, PPARγ agonist–induced edema may be multifactorial in nature, including altered endothelial permeability (Walker et al., 1999; Wagner et al., 2012), sympathetic nervous system activity (Yoshimoto et al., 1997), interstitial ion transport (Hosokawa et al., 1999), and PPARγ-mediated expression of vascular permeability growth factor (Nesto et al., 2004). Nevertheless, more work is needed to elucidate the mechanism(s) involved.
The vasculature represents a potential “target tissue” for TZD-mediated edema since it serves as a direct interface between circulating blood and interstitium, and expression of PPARγ was demonstrated in human endothelial cells (ECs) (Marx et al., 1999; Willson et al., 2001). Indeed, several PPARγ direct and indirect target genes were identified in ECs, encoding proteins relevant to blood pressure regulation and EC permeability, such as nitric oxide production, chemotaxis, apoptosis, and redox signaling (Marx et al., 1999). Focal adhesions between ECs are maintained by focal adhesion kinase (FAK), which normally regulates EC matrix attachment (Romer et al., 2006). FAK normally maintains endothelial adherens junctions, while FAK deletion in mouse ECs disrupts their function and leads to edema (Schmidt et al., 2013). PPARγ agonists decrease FAK expression (Chen et al., 2005), suggesting PPARγ may play a critical role in weakening EC junctions and their major component vascular endothelial (VE)-cadherin (Mehta and Malik, 2006).
To directly examine the hypothesis that EC-expressed PPARγ plays a role in mediating TZD-induced edema in vivo, studies were performed using knockout (KO) mice with targeted disruption of PPARγ in EC (PpargΔEC) (Nicol et al., 2005). Here, we present the first evidence that PpargΔEC mice, versus wild-type (WT) control floxed (Ppargf/f) littermates, are refractory to rosiglitazone-mediated increases in extracellular fluid levels and PVE. Further, both FAK and VE-cadherin are increased among ECs isolated from PpargΔEC versus controls. These results suggest a critical role for EC-expressed PPARγ in mediating TZD-induced edema, which may help human patients treated with these drugs.
Materials and Methods
Animals.
All mice studies were in accordance with protocols approved by the Animal Use and Care Committees of Merck and Queen’s University and conform with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Canadian Council on Animal Care Guidelines. Mice were housed in cages on a 12-hour light/dark cycle with food and water provided ad libitum. Mouse models of endothelial cell–targeted PPARγ deletion (PpargΔEC mice) were generated by crossing our Ppargf/f mice (Akiyama et al., 2002) with Tie2-Cre+ transgenic mice (Kisanuki et al., 2001) as previously described and characterized (Nicol et al., 2005; Kanda et al., 2009). Mice were genotyped using polymerase chain reaction as previously reported (Nicol et al., 2005). Littermates homozygous for the floxed PPARγ gene, but lacking Cre expression, were used as controls. We previously reported characterization of both male and female Ppargf/f or PpargΔEC mice, with very few differences identified, and none of which pertained to ability to respond to a TZD (Nicol et al., 2005). In fact, the only notable sex difference observed was significantly increased heart rate levels among salt-loaded PPARγΔEC KO males compared to similarly treated PPARγf/f WT males, which also trended in that direction for female mice. In light of this and pilot studies showing male mice recapitulated the effects of TZDs on fluid retention, we therefore surmised that male mice would serve as a good model for TZD-induced edema in humans (which occurs to the same extent in both sexes). At 6 weeks of age, male mice were placed on a high-fat diet (HFD; RD12492, 60 kcal% fat, 20 kcal% carbohydrate, and 20 kcal% protein; Research Diets, New Brunswick, NJ) for 12 weeks.
Rosiglitazone Treatment.
Rosiglitazone was incorporated into the diet (D12492; Research Diets) at a level of 100 mg/kg diet. Mice were fed rosiglitazone-formulated or vehicle control diet for a period of 14 days ad libitum.
Plasma Glucose and Insulin Measurements.
Blood taken from the tail vein was collected in heparinized capillary tubes (Clay Adams SurePrep heparinized capillary tubes; Becton Dickinson and Co., Sparks, MD) and centrifuged at 11,500g for 10 minutes to separate plasma. Plasma glucose (Autokit Glucose; Wako Diagnostics, Richmond, VA) and insulin levels (Ultrasensitive rat insulin enzyme-linked immunosorbent assay; ALPCO Diagnostics, Salem, NH) were determined according to protocols provided with the kits.
Bioelectrical Impedance Analysis for Determination of Extracellular Fluid Volume.
Twenty-four hours following the final dose, mice were anesthetized with ketamine (85 mg/kg, i.m.) and xylazine (10 mg/kg, i.m.). Animals were positioned on a nonconductive surface in dorso-lateral recumbency, and extracellular fluid volume was determined by bioelectrical impedance analysis, following procedures described by the manufacturer (Hydra ECF/ICF Impedance Analyzer Model 4; Xitron, San Diego, CA) and by Quirk et al. (1997). A tetrapolar impedance monitor was used to measure impedance, and hence the total body water, over a frequency range of 5 kHz to 1 MHz. Source electrodes (1 cm × 26G stainless steel needles) were inserted 5 mm subcutaneously. Detector electrodes were inserted along the midline at the anterior point of the sternum and the anterior point of the penis. The distance between electrodes was measured and included for data modeling.
Plasma Volume Measurement.
After determination of extracellular fluid volume, plasma volume was measured in anesthetized animals using a dye dilution technique following methods described previously (Belcher and Harriss, 1957), with minor modifications. Evans blue dye solution (25 mg/ml in physiologic saline) was filtered through a 0.22-μm filter prior to injection into a jugular vein. Twenty minutes after injection, a heparinized blood sample (2 ml) was withdrawn from the descending aorta. Plasma was separated by centrifugation of the blood at 1100g for 15 minutes; samples were kept at −80°C until assayed. Absorbance of the thawed plasma was read at 620 nm, and plasma Evans blue dye concentrations were calculated according to a standard curve generated by a serial dilution of the 25-mg/ml Evans blue dye saline solution. Plasma volume was calculated by using the dilution factors of Evans blue as follows:

Tissue Water Content Measurement.
Immediately after euthanasia, epididymal fat pad, pararenal fat pad, inguinal fat pad, and gastrocnemius muscle were removed and weighed, and placed in individual glass scintillation vials. Weighed tissues were subjected to a vacuum-applied Speed Vac centrifugation/drying process over a 24-hour period. After this drying process, tissues were weighed again to calculate individual tissue water content.
Quantitation of Enlarged Capillaries in White Adipose Tissue.
At necropsy, adipose tissue was collected from epididymal and inguinal depots. Samples of white adipose tissue weighing approximately 1 g were fixed by immersion in several changes of 2% freshly prepared paraformaldehyde (in 0.1 M phosphate buffer, pH 7.3) for 48 hours at 4°C. Following fixation, the tissue was dehydrated through graded ethanol, exchanged into xylene, and embedded in paraffin using a Leica TP1020 embedding system (Leica Biosystems Inc, Buffalo Grove, IL). Paraffin sections (8 μm) were subsequently cut on a Leica RM2155 microtome, transferred to glass slides, and stained with hematoxylin and eosin. The number of enlarged capillaries on each slide was quantitated in a blinded fashion as follows: each section was first examined on a Zeiss Axioplan 2 microscope (Carl Zeiss, Oberkochen, Germany) equipped with a digital stage using a 5× objective and a red filter to preclude bias from observing red blood cells (RBCs), which still allowed easy visualization of the tissue. Using the digital stage controls, 10 widely separated locations scattered across the section were marked. This marking ensured that tissue was present and cells at each location were well preserved adipocytes free of mechanical defects, and not large blood vessels, lymph nodes, or connective tissue. After marking 10 locations, each was then re-examined in detail under white-light illumination using a 40× objective. Once the stage was relocated to the marked locations, the stage was not moved in the x-y plane, although focus was adjusted as needed. The number of enlarged capillaries observed in each 40× field was then recorded. For counts, enlarged capillaries were defined as any capillary containing a cluster of three or more RBCs in each of the 10 sections. Capillaries in which the RBCs lined up in a single file were most likely of normal size and were not counted as enlarged.
Immunofluorescent Staining.
Formalin-fixed, paraffin-embedded lung tissue blocks from (n = 3) untreated Ppargf/f and PpargΔEC mice were sectioned into 5-μm slices, mounted on slides, and incubated at 55°C overnight. Samples were deparaffinized and rehydrated by washing in the following: xylene, 4 minutes; xylene, 4 minutes; xylene, 4 minutes; 100% ethanol, briefly; 85% ethanol, briefly; 70% ethanol, briefly; ddH2O, 4 minutes. Slides were placed in 1:10 sodium citrate buffer solution at 95°C for 20 minutes, then trypsinized with 1× trypsin (Sigma-Aldrich, St. Louis, MO) for 20 minutes at 37°C. After washing, the slides were placed in Triton X/Tris-buffered saline (TBS) buffer solution, followed by a 30-minute incubation period in 5% bovine serum albumin (BSA) block in TBS. After washing, primary antibody for VE-cadherin (ALX-803-305-C100; 1:200; Enzo Life Sciences, Farmingdale, NY) was applied in a 5% BSA solution for 3.5 hours at room temperature. Slides were rinsed with TBS and then incubated in fluorescein isothiocyanate (1:500; Santa Cruz Biotechnology, San Diego, CA) and Alexa Fluor 594 (1:500; Invitrogen, Carlsbad, CA) fluorescent-conjugated secondary antibodies in 5% BSA for 15 minutes at room temperature. After a final rinsing regimen, tissues were cover slipped with mounting medium containing 4′,6-diamidino-2-phenylindole stain (Vectashield, Vector Laboratories, Burlingame, CA). Fluorescence was detected and quantitated using a Quorum WaveFX-X1 spinning disk confocal system (Quorum Technologies, Guelph, ON, Canada) and MetaMorph Offline (64-bit, version 7.7.0.0, Molecular Devices, San Jose, CA).
Endothelial Cell Isolation.
ECs were isolated from lung samples collected from (n = 3) untreated Ppargf/f and PpargΔEC mice according to an established protocol (Kobayashi et al., 2005). In brief, lungs were collected at necropsy, minced finely, digested with collagenase A, and washed repeatedly. ECs were selected using dynabeads (Life Technologies, Burlington, ON, Canada) conjugated to PECAM-1 antibody (Santa Cruz Biotechnology) following the manufacturer’s instructions. The dynabeads were washed four times with 4 ml of 10% fetal bovine serum in Dulbecco’s modified Eagle’s medium to ensure purity of the EC population, and then trypsinized to remove the cells from the beads. Total RNA was collected immediately following cell isolation according to the TRIzol method. In brief, cells were suspended in TRIzol (Invitrogen) for 5 minutes, then chloroform was added for 3 minutes. After centrifugation, RNA was extracted from the aqueous layer using isopropanol (10 minutes), 75% ethanol (5 minutes), and resuspended in RNase-free water.
Quantitative Real-Time Polymerase Chain Reaction Assay.
Following UV-spectrophotometric analyses (A260/280) to determine purity and concentrations, RNA samples were converted to cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). cDNA concentrations were quantified as described earlier for RNA and combined with iQ SYBR Green mix (Bio-Rad), autoclaved commercially available ddH2O, and PrimePCR Assay primers (cat.# 10025636; Bio-Rad) to evaluate FAK (PTK2) expression levels. Assays were performed on an iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad) thermocycler programmed with the following conditions: 5-minute 95°C hot-start followed by 50 cycles of 95°C for 15 seconds, 65°C for 15 seconds, and 72°C for 30 seconds.
Statistical Analysis.
All values are presented as means ± S.E. Statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA). Multiple group comparisons were assessed using a two-way analysis of variance followed by Bonferroni post hoc tests, with a P value <0.05 considered statistically significant.
Results
PpargΔEC Mice Are Healthy, Viable, and Able to Gain Weight Normally on an HFD.
PpargΔEC mice are viable, appear healthy, and exhibit no gross abnormalities. After 12 weeks on an HFD, PpargΔEC mice did not differ from Ppargf/f control littermates in terms of body weight (PpargΔEC mice 41.4 ± 0.9 g vs. PPARγf/f 42.7 ± 0.9 g) or food intake (PpargΔEC 17.9 ± 0.4 kcal/day vs. Ppargf/f 17.6 ± 0.5 kcal/day, based on RD12492 diet). Baseline lean, fat, and fluid mass in PpargΔEC mice were also similar to Ppargf/f controls as measured by quantitative NMR (Fig. 1). Others have reported an attenuation in rosiglitazone-induced body weight gain in collecting duct-targeted PPARγ-null mice versus control mice, presumably due, in part, to reduced fluid retention (Guan et al., 2005). In this study, there was a tendency for rosiglitazone-treated Ppargf/f mice to gain weight relative to corresponding vehicle-treated mice; however, this trend was not statistically significant (Supplemental Fig. 1). This apparent discrepancy in results likely reflects the difference in the baseline body weights of the mice prior to treatment [∼42 g in this study vs. ∼29 g in the Guan et al. (2005) study] since rosiglitazone is more likely to promote weight gain in leaner mice.

High-fat diet effect on body mass composition of Ppargf/f and PpargΔEC mice. Quantitative nuclear magnetic resonance imaging (qNMR) was performed on Ppargf/f and PpargΔEC mice following 12-week treatment on an HFD. Values are presented as a percentage of total body weight.
EC-Expressed PPARγ Is Not Required for TZD-Mediated Antidiabetic Efficacy.
Prior to rosiglitazone treatment, the baseline levels of ambient plasma glucose and insulin were unchanged in PpargΔEC mice relative to Ppargf/f mice on an HFD (Fig. 2). Rosiglitazone treatment of Ppargf/f mice resulted in a dramatic reduction in plasma insulin but not glucose levels (Fig. 2), consistent with the results of other studies in C57B6 mice treated with TZDs (Gensch et al., 2007; Liu et al., 2007). In PpargΔEC mice, rosiglitazone also normalized plasma insulin levels to a similar extent as in Ppargf/f, with no effect on glucose levels (Fig. 2), suggesting that EC-expressed PPARγ is not required for TZD-mediated improvement in insulin sensitivity.

Rosiglitazone effects on plasma glucose and insulin levels in Ppargf/f and PpargΔEC mice. Plasma glucose (A) and insulin (B) values were determined in Ppargf/f and PpargΔEC mice prior to and following vehicle or rosiglitazone treatment for 14 days, as described in Materials and Methods. Values represent the mean + S.E. *Significantly different compared with respective vehicle controls (P < 0.05).
A histologic analysis of epididymal and inguinal white adipose tissue (eWAT and iWAT) depots in control mice treated with rosiglitazone revealed the presence of pockets of smaller, more multilocular adipocytes (Fig. 3, A and B). Such changes are consistent with a more “activated” adipocyte phenotype in response to PPARγ agonism and have been described in other rodent models (Okuno et al., 1998; de Souza et al., 2001). Similar changes in the size and arrangement of adipocytes were also observed in eWAT and iWAT of PpargΔEC mice treated with rosiglitazone (Fig. 3, C and D). No significant differences were observed in assessed tissue weights (Supplemental Fig. 1). It was suggested that a TZD-induced shift toward smaller adipocytes improves insulin resistance by decreasing the proportion of larger adipocytes that secrete elevated levels of free fatty acids (Okuno et al., 1998). Consistent with this notion, the appearance of multilocular adipocytes coincides with similar insulin lowering in both PpargΔEC and Ppargf/f mice upon rosiglitazone treatment. In addition, the presence of multilocular adipocytes in rosiglitazone-treated PpargΔEC may serve as an internal control demonstrating that PPARγ expression in adipocytes is unaffected by EC-targeted disruption of PPARγ.
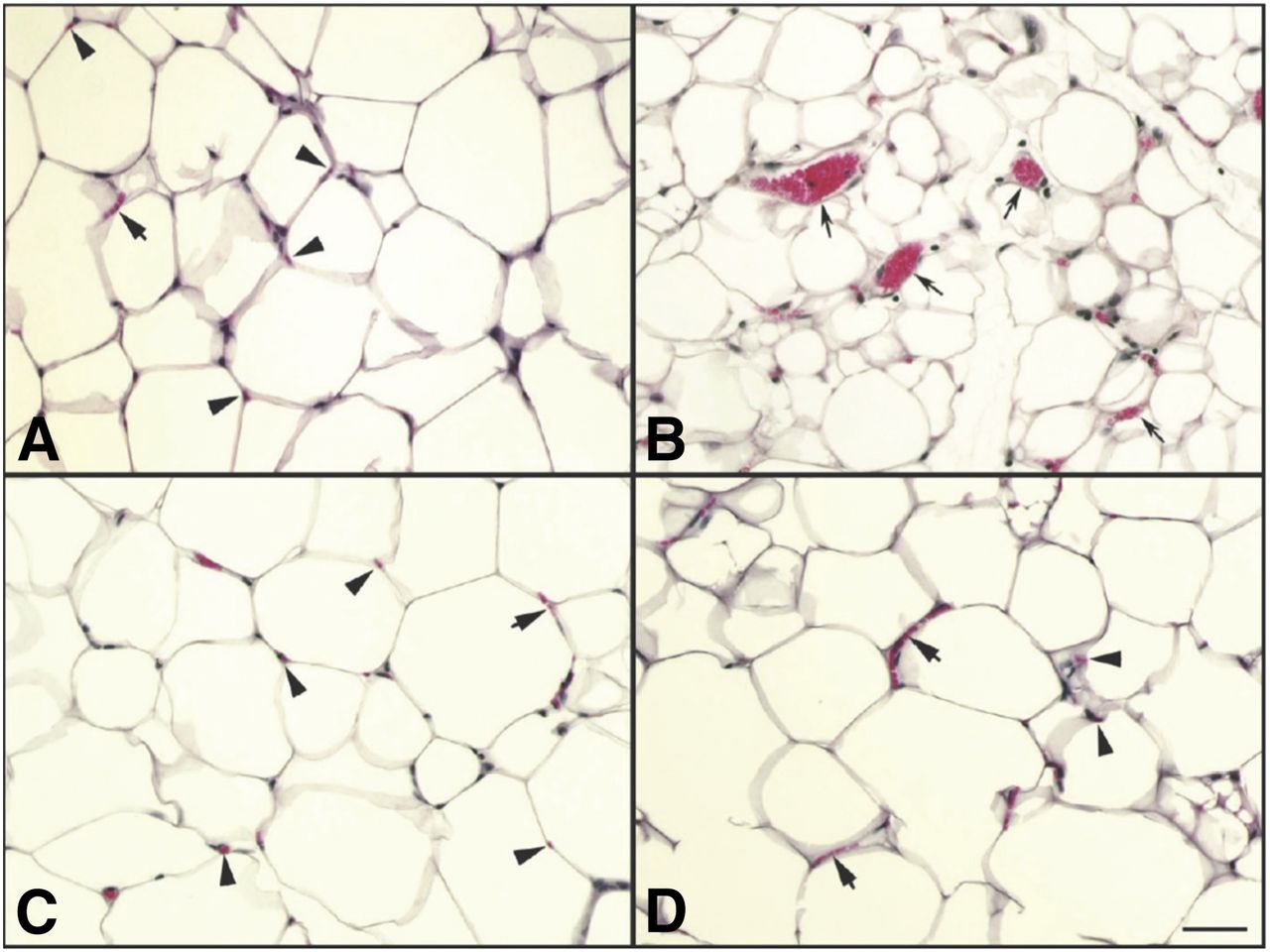
Histologic analysis of epididymal white adipose tissue from Ppargf/f and PpargΔEC mice. Formalin-fixed, paraffin-embedded samples from each treatment group were assessed as described in Materials and Methods. Vehicle-treated Ppargf/f mice (A), rosiglitazone-treated Ppargf/f mice (B), vehicle-treated PpargΔEC mice (C), and rosiglitazone-treated PpargΔEC mice (D). The bar in the lower right of (D) is 50 μm long. Arrowheads in (A), (C), and (D) indicate single red blood cells in a normal capillary cut in cross section. Short arrows in (A), (C), and (D) point to multiple red blood cells lined up in single file in a normal capillary laying in the plane of the section. Long (concave) arrows in (B) point to abnormally large capillaries observed only in eWAT from rosiglitazone-treated Ppargf/f mice.
EC-Expressed PPARγ Plays a Critical Role in TZD-Induced Capillary Vasodilation.
The hypotensive effects of TZDs in humans and preclinical species are well documented (Pershadsingh et al., 1993; Buchanan et al., 1995; Ogihara et al., 1995; Walker et al., 1999). Although the mechanism(s) for TZD-induced blood pressure lowering is not entirely clear, a reduction in total peripheral resistance may be an important component of this response since TZDs cause significant capillary vasodilation in preclinical species (Buchanan et al., 1995; Kotchen et al., 1996; Ghazzi et al., 1997; Song et al., 1997; Kawasaki et al., 1998). PPARγ activation with TZDs also decreases the pressure-induced platelet aggregation that leads to hypertension (Rao et al., 2014). Hypertension is also driven by the renin-angiotensin-aldosterone system, and multiple groups have shown that PPARγ ligands reduce blood pressure by inhibiting the renin-angiotensin-aldosterone system (Sugawara et al., 2012). Multiple PPARγ ligands decrease mRNA expression of the angiotensin II type 1 receptor expression (Sugawara et al., 2001). Additionally, PPARγ agonists inhibit angiotensin-induced aldosterone production/secretion by suppressing aldosterone synthase (CYP11B2) (Uruno et al., 2011). In one study using human subjects, it was concluded that rosiglitazone does not affect vasodilation (Rennings et al., 2006); however, this finding is not supported by the majority of the literature.
In the current study, Ppargf/f mice treated with rosiglitazone exhibited a clear increase in the number of enlarged capillaries in both eWAT and iWAT depots versus vehicle treatment (Fig. 4B). Strikingly though, the effects of rosiglitazone on capillary vasodilation (Fig. 3, C and D) in white adipose tissue depots of control mice were noticeably absent in PpargΔEC mice. Interestingly, these vascular effects of rosiglitazone appear to be tissue-specific since rosiglitazone treatment did not affect capillary vasodilation within skeletal muscle tissue (data not shown). The lack of rosiglitazone-induced capillary expansion in the white adipose tissue of PpargΔEC mice may also help to explain the resistance to TZD-induced blood pressure lowering shown previously in the same mouse model (Nicol et al., 2005). Together, these results indicate that EC-expressed PPARγ is necessary for mediating the vasodilatory effects of rosiglitazone in adipose tissue and may provide mechanistic insight into blood pressure regulation by TZDs.

Quantitation of enlarged capillaries in epididymal and inguinal white adipose tissue. Epididymal (A) and inguinal (B) adipose tissue were fixed, dehydrated, paraffin embedded, and stained with hematoxylin and eosin. Enlarged capillaries were from 10 widely separated locations of preserved adipocytes, not large blood vessels, lymph nodes, or connective tissue. *Significantly different compared with respective vehicle controls (P < 0.05). VEH, vehicle; ROSI, rosiglitazone.
EC PPARγ Plays a Critical Role in TZD-Induced Extracellular Fluid and Plasma Volume Expansion.
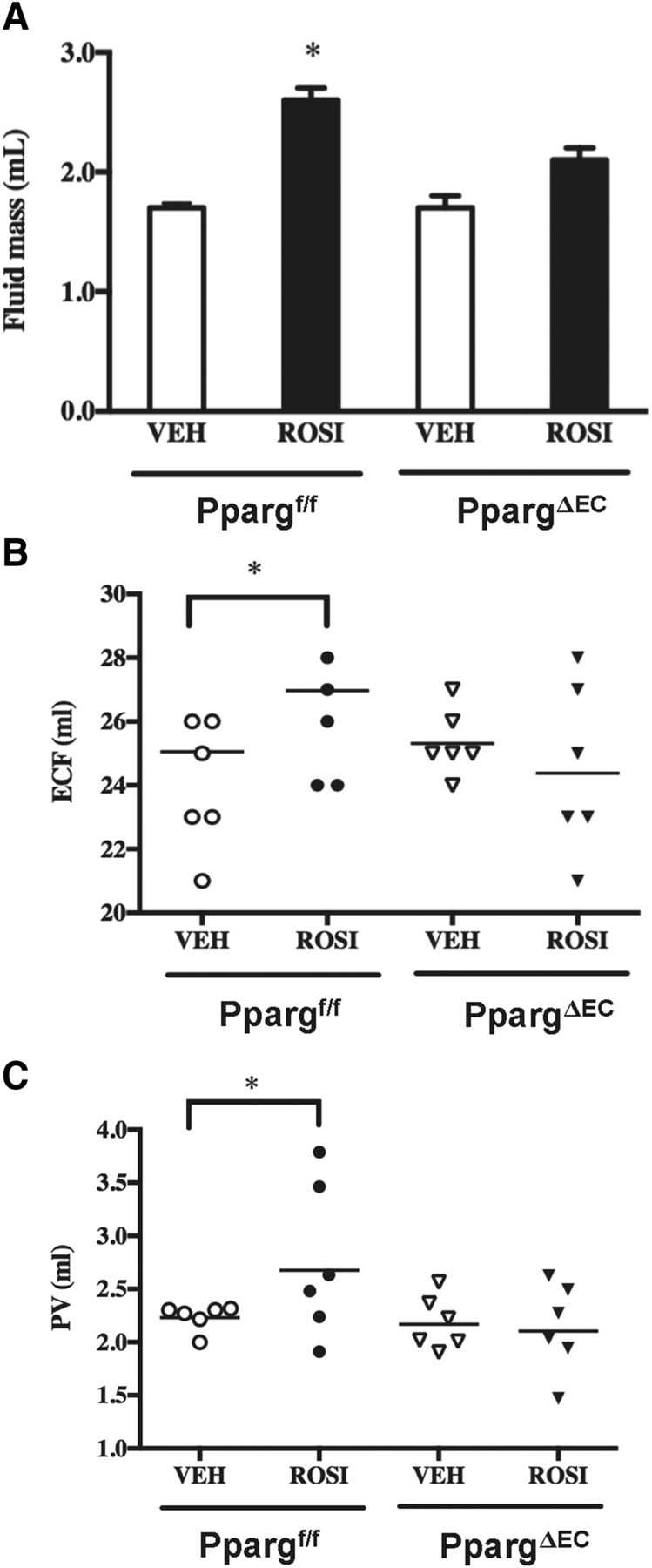
To address whether the observed vascular changes in WAT following rosiglitazone treatment could ultimately impact systemic fluid dynamics, the effect of rosiglitazone on multiple surrogate markers of fluid accumulation was examined in PpargΔEC mice. First, the results of quantitative nuclear magnetic resonance imaging measurement showed that while the EC-targeted deletion of PPARγ did not affect whole-body fluid mass, rosiglitazone induction of total fluid mass in control mice was attenuated in PpargΔEC mice (Fig. 5A).
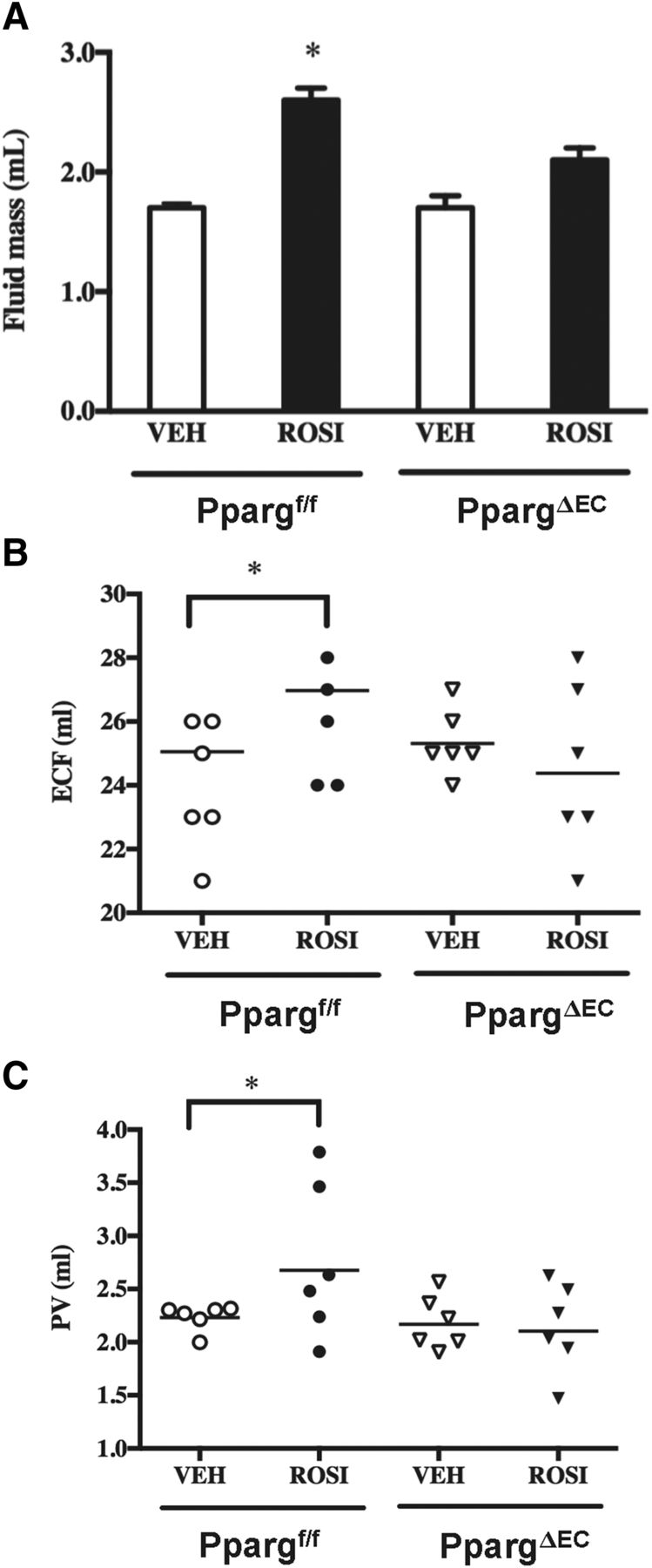
Role of endothelial PPARγ expression in measures of rosiglitazone-mediated edema. (A) Quantitative nuclear magnetic resonance imaging was performed on Ppargf/f and PpargΔEC mice following vehicle (VEH) or rosiglitazone (ROSI) treatment as described earlier. Values represent absolute fluid mass in milliliters. (B) Extracellular fluid volume in Ppargf/f and PpargΔEC mice following vehicle or rosiglitazone treatment as determined by bioimpedance measurements. (C) Plasma volume (PV) in Ppargf/f and PpargΔEC mice following vehicle or rosiglitazone treatment as determined by the Evan’s blue assay. All values are reported as the mean + S.E. *Significantly different compared with respective vehicle controls (P < 0.05).
Next, bioelectrical impedance, a term used to describe the response of a living organism to an externally applied electric current, was examined in both vehicle- and rosiglitazone-treated PpargΔEC and Ppargf/f mice. Measurement of the opposition to the flow of that electric current through the tissues has proven useful as a noninvasive method for quantifying extracellular fluid (ECF) (Quirk et al., 1997). Our results show that while basal levels of ECF are not significantly different in vehicle-treated PpargΔEC mice versus Ppargf/f controls, PpargΔEC mice are almost completely resistant to the rosiglitazone induction of ECF observed in Ppargf/f mice (Fig. 5B).
The Evans blue technique is widely used to determine plasma volume in humans, dogs, and rats (Merrill and Cowley, 1987; Iwasaki et al., 2004; Ross and Idah, 2004). Using this technique, PpargΔEC mice have normal plasma volume levels that are not significantly different from control mice (Fig. 5C). However, the response to rosiglitazone was markedly attenuated compared with Ppargf/f mice, in which a significant elevation in plasma volume was observed (Fig. 5C). Taken together, these data indicate that EC PPARγ plays a central role in regulating plasma volume and ECF following TZD treatment.
PpargΔEC Mice Are Resistant to Rosiglitazone-Induced Fluid Accumulation in White Adipose Tissue.
Following rosiglitazone treatment, tissue biopsies were collected from eWAT, iWAT, and retroperitoneal white adipose tissue and skeletal muscle. Tissue weights were recorded prior to and after lyophilization of the tissue biopsies and normalized to baseline “wet” tissue weights to determine tissue water content. Rosiglitazone-treated control mice exhibited markedly increased water content in each of the WAT depots examined, but not in skeletal muscle (Fig. 6A-D). On the other hand, water content in both skeletal muscle and white adipose tissue remained unchanged in rosiglitazone-treated PpargΔEC mice versus vehicle-treated controls (Fig. 6A-D). Notably, the appearance of capillary vasodilation and elevated tissue water content coincide in terms of tissue specificity (white adipose tissue but not skeletal muscle). Based on this, the possibility exists that arterial fluid more readily crosses the endothelium of enlarged capillaries within white adipose tissue, although capillary permeability was not specifically examined in this study. The combined data imply that TZD-induced fluid accumulation occurs in a tissue-specific manner and requires a functional PPARγ receptor in ECs.

Tissue water content in vehicle (VEH)- or rosiglitazone (ROSI)-treated Ppargf/f and PpargΔEC mice. Water content in epididymal (A), inguinal (B), and retroperitoneal white adipose tissue (rWAT) (C) and skeletal muscle (D) in VEH- or ROSI-treated Ppargf/f and PpargΔEC mice was assessed following tissue collection at necropsy. The difference between wet tissue weight and dry tissue weight following lyophilization (reflecting absolute water weight) was normalized to wet tissue weight prior to lyophilization. All values are reported as the mean + S.E. *Significantly different compared with respective vehicle controls (P < 0.05).
PPARγ Deficiency in ECs Leads to Alterations in Endothelial Cell Junctions.
Fak mRNA expression was measured in lung endothelial cells isolated from PpargΔEC and Ppargf/f mice. Lung tissue was chosen here due to its high proportion of endothelial cells. Relative to Ppargf/f mice, PpargΔEC mice expressed less than 0.04% Pparg mRNA, normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, in their ECs (P = 0.055) (Fig. 7A). Despite not being statistically significant, this difference is likely biologically significant and is consistent with our previous data demonstrating PPARγ within ECs was disrupted in our mouse model (Nicol et al., 2005). Additionally, Fak mRNA expression, normalized to GAPDH mRNA, was 15-fold higher in PpargΔEC ECs versus controls (P = 0.06) (Fig. 7B). Importantly, this result is consistent with other reports that PPARγ negatively regulates FAK expression (Chen et al., 2005), which may explain the increased sensitivity to TZD-induced edema observed in Ppargf/f mice treated with rosiglitazone.
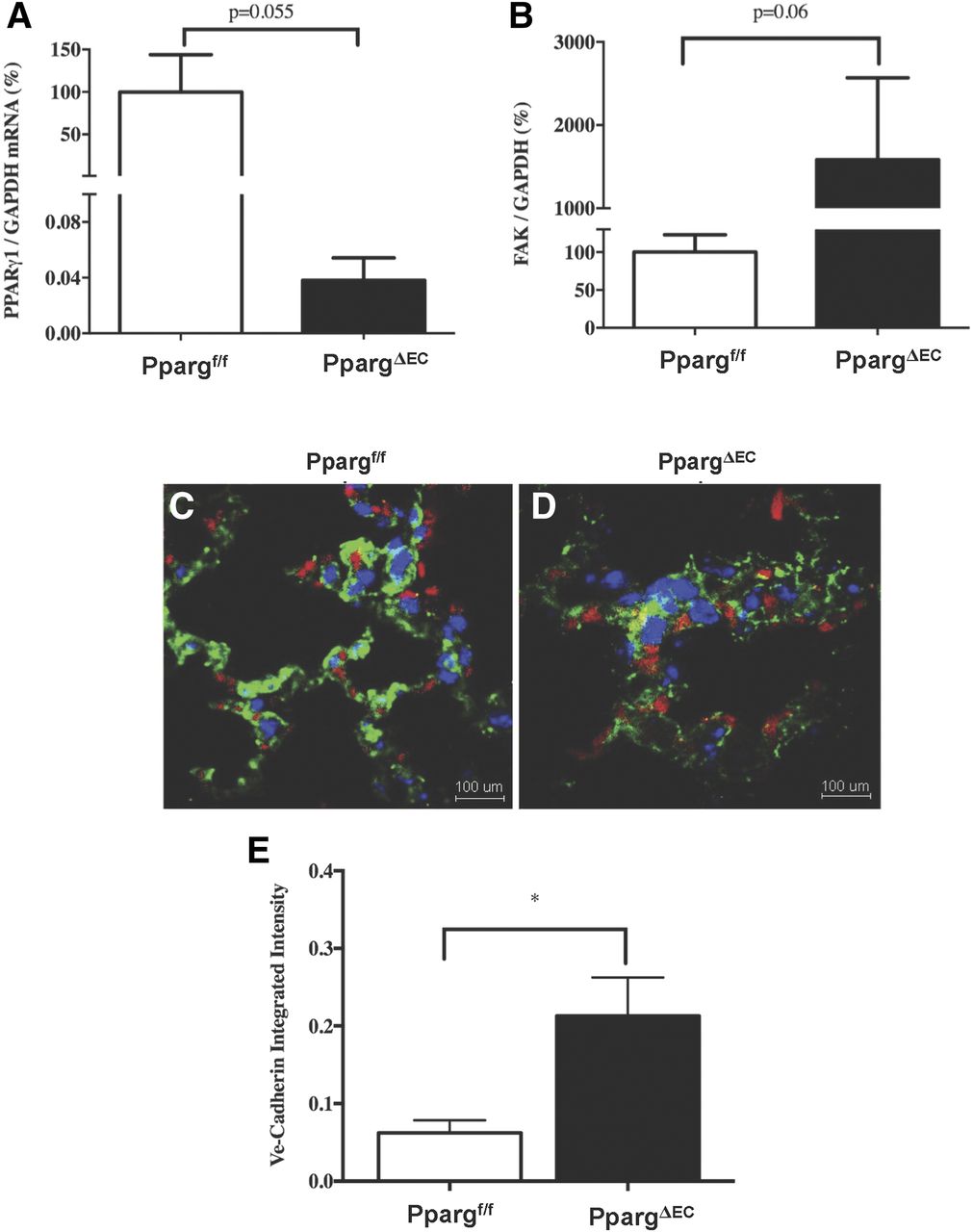
Endothelial expression changes in vehicle- or rosiglitazone-treated Ppargf/f and PpargΔEC mice. (A and B) Analysis of Pparg and Fak mRNAs within isolated lung ECs from (n = 3) untreated Ppargf/f and PpargΔEC mice. All values are calculated relative to the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, and reported as a mean percentage + S.D. of mRNA expression in Ppargf/f mice. (C–E) Immunofluorescent evaluation of VE-cadherin in EC junctions from lung samples of (n = 3) Ppargf/f and PpargΔEC mice. Samples were stained with 4′,6-diamidino-2-phenylindole (blue), PECAM (green), and VE-cadherin (red). (C) Representative image from Ppargf/f lung sample. (D) Representative image from PpargΔEC lung sample. (E) Integrated intensity of VE-cadherin expression in EC junctions of Ppargf/f and PpargΔEC mice. *Significantly different compared with controls (P < 0.05).
Given the increase in Fak expression, immunofluorescence was used to evaluate EC junctions. Formalin-fixed, paraffin-embedded lung samples from (n = 3) PpargΔEC and Ppargf/f mice were cut and stained with PECAM and VE-cadherin to identify EC populations and junctions, respectively. VE-cadherin expression quantified in PECAM+ cells was significantly >3-fold higher in PpargΔEC samples versus Ppargf/f controls (P < 0.05) (Fig. 7, C–E).
Discussion
PPARγ agonists have potent cardiovascular effects including capillary vasodilation, mean arterial blood pressure lowering, and PVE. However, the precise mechanisms by which these effects occur is not entirely clear and may be multifactorial. The results of multiple studies, including both pharmacologic and genetic, suggest that fluid retention mediated by PPARγ in the collecting duct is an important factor that contributes to the development of TZD-induced edema. In addition to the kidney, PPARγ is well expressed in ECs. This study was undertaken with the goal of better understanding what role, if any, PPARγ in the endothelium plays in TZD-induced edema. The side effects of TZDs observed in humans, including PVE, hemodilution, edema, increased adiposity, and weight gain, have been recapitulated in rodent species (Berger and Moller, 2002); thus, preclinical rodent models are useful to help define the in vivo mechanisms causing these effects. Toward this end, PpargΔEC mice were placed on an HFD (a condition that promotes PPARγ agonist–induced PVE) and treated with a prototypical TZD, rosiglitazone.
Our results showed that PpargΔEC mice had similar total body weight gain as well as lean, fat, and fluid mass relative to control littermates on HFD and no significant changes in body or tissue weights following rosiglitazone treatment. PpargΔEC mice also remained responsive to the insulin-lowering effects of rosiglitazone, suggesting that PPARγ in ECs is not critical to overall metabolic phenotype (at least related to diabetes and obesity) under ambient conditions. In response to rosiglitazone treatment, however, a number of interesting phenotypes emerged in the PpargΔEC mice that have relevance to both the regulation of blood pressure and development of edema. With regard to the former, the results of histologic analysis indicate that PpargΔEC mice are refractory to the capillary vasodilatory effects of rosiglitazone in multiple white adipose depots. In addition, rosiglitazone also reduced adipocyte size, an effect that is consistent with previous findings showing that a TZD agonist of PPARγ increased the number of small adipocytes without a change in white adipose tissue mass in obese Zucker rats (Okuno et al., 1998). Based on these observations, one might therefore expect that TZDs do not significantly reduce total peripheral resistance and blood pressure in PpargΔEC mice unless under diabetic conditions, in agreement with a previous report (Nicol et al., 2005). Histologic analysis also revealed that rosiglitazone-induced capillary vasodilation occurs in a tissue-specific manner (white adipose but not in skeletal muscle). It is tempting to speculate that paracrine effects originating from adipocytes, where PPARγ is highly expressed, contribute to vascular remodeling in adipose tissue, a possibility that requires future investigation, for example, by using adipocyte-targeted PPARγ KO mice. An obvious implication of our histologic observations is that TZD-induced capillary vasodilation leads to increased capillary permeability and the development of edema.
Concerns over the potential for TZDs to promote the risk of myocardial infarction (Nissen and Wolski, 2007; Kung and Henry, 2012) prompted the FDA to require black box label warnings on Avandia in 2007 (Starner et al., 2008). However, the FDA lifted restrictions in 2013 after re-evaluating the 2009 RECORD clinical trial (a 6-year, open-label, randomized control trial), which did not show heart infarct risks associated with the drug (Home et al., 2009; http://www.fda.gov/Drugs/DrugSafety/ucm376389.htm). In 2015, the FDA further removed the Risk Evaluation and Mitigation Strategy requirements for rosiglitazone-containing medicines after continuous monitoring indicated no new pertinent safety information (http://www.fda.gov/Drugs/DrugSafety/ucm476466.htm). Collectively, our data are the first to show that normal PPARγ expression suppresses EC-specific FAK expression, which in turn regulates EC junctions and vascular permeability. This suggests a mechanistic link that may explain the rosiglitazone-induced edema demonstrated by Ppargf/f mice but not PpargΔEC mice (Fig. 8). By negatively regulating FAK, PPARγ in ECs may cause the destabilization of EC junctions. This is further suggested by the reduction of VE-cadherin in lung EC junctions in Ppargf/f compared with PpargΔEC mice. Indeed, a consensus statement from the American Heart Association and the American Diabetes Association posited that TZDs may interact synergistically with insulin to cause arterial vasodilation, leading to sodium reabsorption with a subsequent increase in extracellular volume, thereby resulting in peripheral edema in humans (Nesto et al., 2004). It is also suggested that the combination of decreased total peripheral resistance and expanded plasma volume is sufficient to cause an increase in interstitial fluid retention (Chen et al., 2005). Rosiglitazone-induced arterial vasodilation was accompanied by increased total body fluid mass as determined by quantitative nuclear magnetic resonance imaging, increased ECF, elevated tissue water content in white adipose tissue, as well as plasma volume expansion in Ppargf/f mice but not PpargΔEC mice, suggesting a direct PPARγ-dependent effect.

Summary model of PPARγ-dependent edema mechanism. Left panel, Rosiglitazone (ROSI)-mediated activation of PPARγ signaling in ECs suppresses FAK, leading to direct or indirect decreases in VE-cadherin expression and EC adherens junctions, allowing for increased edema. Right panel, Loss of PPARγ expression/signaling abrogates FAK repression and enhances VE-cadherin expression and adherens junctions between ECs, thereby minimizing edema.
In summary, this study shows that plasma volume expansion and increased extracellular fluid volume (i.e., edema) are mediated by rosiglitazone acting directly on receptors present in the vascular endothelium. The combined effects of a PPARγ agonist on both renal and vascular PPARγ thus appear to contribute to the induction of edema. Given that both rosiglitazone and pioglitazone are known to cause PPARγ-induced edema, the results of this study are likely broadly applicable to both rosiglitazone and pioglitazone treatment of human type 2 diabetes patients. The involvement of PPARγ expressed in the hematopoietic system cannot be ruled out as a confounder in mediating the effects of rosiglitazone, since Tie2 expression is reported in multiple hematopoietic cell types, including B cells and T cells (Constien et al., 2001). Modulation of inflammatory signaling may influence factors such as capillary permeability and vascular tone, and remains to be investigated.
From a therapeutic standpoint, the identification of selective modulators of PPARγ that maintain robust antidiabetic efficacy with reduced PVE in rodents suggests that these novel ligands may have effects on ECs that are distinct from those of TZDs and other full agonists of PPARγ. Further studies that focus on PPARγ activity in ECs and kidney should help to clarify the mechanisms responsible for the improved therapeutic window of selective modulators of PPARγ in preclinical species, and may lead to the identification of potential biomarkers of PPARγ agonist–induced edema.
Acknowledgments
We acknowledge Sarah Nersesian from Designs that Cell for illustrations (https://designsthatcell.wixsite.com/home). There are no conflicts of interest to declare.
Authorship Contributions
Participated in research design: Akiyama, Skelhorne-Gross, Gonzalez, Berger, Nicol.
Conducted experiments: Akiyama, Skelhorne-Gross, Lightbody, Rubino, Shi, McNamara, Sharma, Zycband, Liu, Woods, Chang, Nicol.
Contributed new reagents or analytic tools: Gonzalez, Nicol.
Performed data analysis: Akiyama, Skelhorne-Gross, Lightbody, Rubino, Shi, McNamara, Sharma, Zycband, Liu, Woods, Chang.
Wrote or contributed to the writing of the manuscript: Akiyama, Skelhorne-Gross, Lightbody, Gonzalez, Berger, Nicol.
Footnotes
- Received May 28, 2018.
- Accepted December 6, 2018.
↵1 T.E.A. and G.E.S.-G. contributed equally to this work.
This work was supported by funding from Merck Research Laboratories to T.E.A., L.A.M., N.S., E.I.Z., H.L., J.W.W., C.H.C., and J.P.B., and the Canadian Breast Cancer Foundation, Ontario Chapter [#369649] to C.J.B.N. This research was supported in part by the Intramural Research Program of the National Institutes of Health [National Cancer Institute]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
This article was prepared while T.E.A. was employed at Merck & Co. The opinions expressed in this article are the author’s own and do not reflect the view of the Food and Drug Administration, the Department of Health and Human Services, or the United States government.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- BSA
- bovine serum albumin
- EC
- endothelial cell
- ECF
- extracellular fluid
- eWAT
- epididymal white adipose tissue
- FAK
- focal adhesion kinase
- FDA
- Food and Drug Administration
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HFD
- high-fat diet
- iWAT
- inguinal white adipose tissue
- PPARγ
- peroxisome proliferator-activated receptor γ
- PpargΔEC
- Pparg EC-targeted knockout
- Ppargf/f
- Pparg-floxed wild-type control
- PVE
- plasma volume expansion
- ROSI
- rosiglitazone
- RBC
- red blood cell
- TBS
- Tris-buffered saline
- TZD
- thiazolidinedione
- VEH
- vehicle
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}