


Abstract
In the 1980s, researchers used binding studies to show that there is a melatonin binding site in addition to the receptors described previously. It was first termed ML2 and then, in 1999, MT3. Purification efforts led to its identification as quinone reductase 2. Several lines of evidence support the notion that MT3 is the same as quinone reductase 2, including the detection and characterization of MT3 whenever quinone reductase 2 was added to various systems under various conditions. This evidence is discussed in this review, which summarizes the results of relevant cellular and animal experiments. The recent discovery that the quinone reductase 2 enzyme can be partly membrane-associated may unite the current body of evidence and allow us to conclude definitively that the third melatonin binding site, MT3, is indeed quinone reductase 2.
Introduction
Melatonin is a small neurohormone derived from tryptophan (Jockers et al., 2016). It has multiple activities at pharmacologic concentrations (µM and greater) and could act as a free radical scavenger. At physiologic concentrations (nM and lower), melatonin helps mediate the perception of day and night rhythms. Indeed, it is produced by the pineal gland mainly at night; thus, it relays circadian rhythms to the peripheral organs. Reports recently added a list of locations (mitochondria, mostly) at which melatonin could be produced (Suofu et al., 2017) or alternative sources (vegetables) from which it could be ingested (Tan et al., 2014). Melatonin interacts with several target proteins. Two such targets are the receptors MT1 (Reppert et al., 1994) and MT2 (Reppert et al., 1995), mammalian G-protein–coupled receptors (GPCRs) that were identified two decades ago. Another target is Mel1c, a GPCR that is functional in only amphibians and birds (Ebisawa et al., 1994). Notably, Mel1c evolved into gpr50 in most mammals, losing its capacity to bind melatonin (Dufourny et al., 2008). Although a mammal, the platypus (Ornithorhynchus anatinus) expresses functional Mel1c/gpr50 (Gautier et al., 2018b). The role of gpr50 in mammals remains a matter of debate, as it has been implicated in an inhibitory dimerization with MT1 (Levoye et al., 2006) while playing an important role in adaptive thermogenesis and torpor (Bechtold et al., 2012), as well as identified as a TGFβ receptor type 1 coreceptor (Wojciech et al., 2018) with potential impact on cancer development (Ahmad et al., 2018). Another melatonin binding site, MT3, which was known as ML2 before 1999, was described by Duncan et al. (1988) and is the subject of the present review. Lastly, a nuclear melatonin receptor has been described and debated over the years; despite the controversy, it seems clear that melatonin interacts with another protein target (or possibly with its signaling pathway). Owing to the large amount of publications recently linking melatonin and Nrf2, this protein might very well be a major or one of the main causes for melatonin’s antioxidant effects (Zhang et al., 2018). Recent reviews have summarized the pharmacology (Cecon et al., 2018) and the signaling pathways (Oishi et al., 2018) of MT1 and MT2.
A recent review of melatonin receptors (Jockers et al., 2016) and an article on MCA-NAT and its effect on intraocular pressure (Martinez-Aguila et al., 2016) both mentioned that additional data are needed to validate that MT3 is quinone reductase 2. To address this viewpoint, it is useful to summarize what is known about the identity and characteristics of MT3.
History of MT3 Binding Site
The MT3 melatonin binding site was originally described in the Syrian hamster brain, in 1988, when melatonin receptors had not yet been cloned. Hamsters have been used extensively as an animal model to study the photoperiodic effects of melatonin. Duncan et al. (1988) reported a binding site at which melatonin association and dissociation were so rapid that experiments had to be conducted at 0°C, which is rare in receptor research. Their data clearly showed that this binding site was membrane-associated. The ligand pharmacologic profile was different in hamsters from other species, particularly compared with the chicken (although melatonin binding in chickens was potentially due to Mel1c) and bovine brain (in which binding was potentially due to MT1 and MT2). A subsequent study by this group further characterized this binding site (Duncan et al., 1989). It was unknown whether hamster MT1 contributed to this binding profile. It would take 8 more years for Reppert’s group to clone the melatonin MT1 receptor in the Siberian hamster after they had cloned MT1 and MT2 in humans and in other mammals (Reppert et al., 1994, 1995). This group further showed that Syrian hamsters were a natural knockout organism for this MT2 receptor subtype (Weaver et al., 1996). Our group recently confirmed the pharmacologic characteristics of the hamster MT1 receptor, as well as the difficulty of obtaining the MT2 receptor from European hamsters (Gautier et al., 2018a), although it is probably not knocked out in this subspecies of hamster.
A summary of the MT3 binding characteristics and ligand profile can be found in Dubocovich (1988). Based on what was known at that time, this binding site is clearly different from the MT1 and MT2 (Table 1) (Dubocovich, 1995). Specifically, two compounds were used to describe this binding site: prazosin, the well known adrenoceptor ligand, and the newly described 5-methoxycarbonylamino-N-acetyltryptamine (MCA-NAT) (Molinari et al., 1996). Pickering and Niles (1990, 1992) reported that the compound prazosin could be used to differentiate MT3 from the other melatonin receptors and binding sites. Specifically, prazosin showed a high affinity (9 nM) for MT3 but not for the cloned human MT1 or MT2 receptors (Paul et al., 1999) (Table 1). The pharmacologic profile of MT3 was therefore distinct, based on its interactions with prazosin, a highly selective α1-adrenergic antagonist, as well as based on the newly described compound, MCA-NAT, also called GR135531 (Dubocovich, 1995). The pharmacologic profile of the binding site was definitely different from the profiles of the cloned MT1 and MT2 receptors (Paul et al., 1999; Nosjean et al., 2001). Indeed, the consensus in the field was that the characteristics of this binding site were unlike those of the other melatonin GPCRs. We and others repeated these experiments and confirmed that the site equilibrated extremely rapidly and that the melatonin association half-life was in the 1- to 10-second range (Paul et al., 1999). In contrast, the melatonin GPCRs showed equilibration in the hour range at room temperature (Table 1).
Characteristics of the melatonin binding sites MT1, MT2, and MT3
This table is loosely based on the one from Dubocovich (1995).
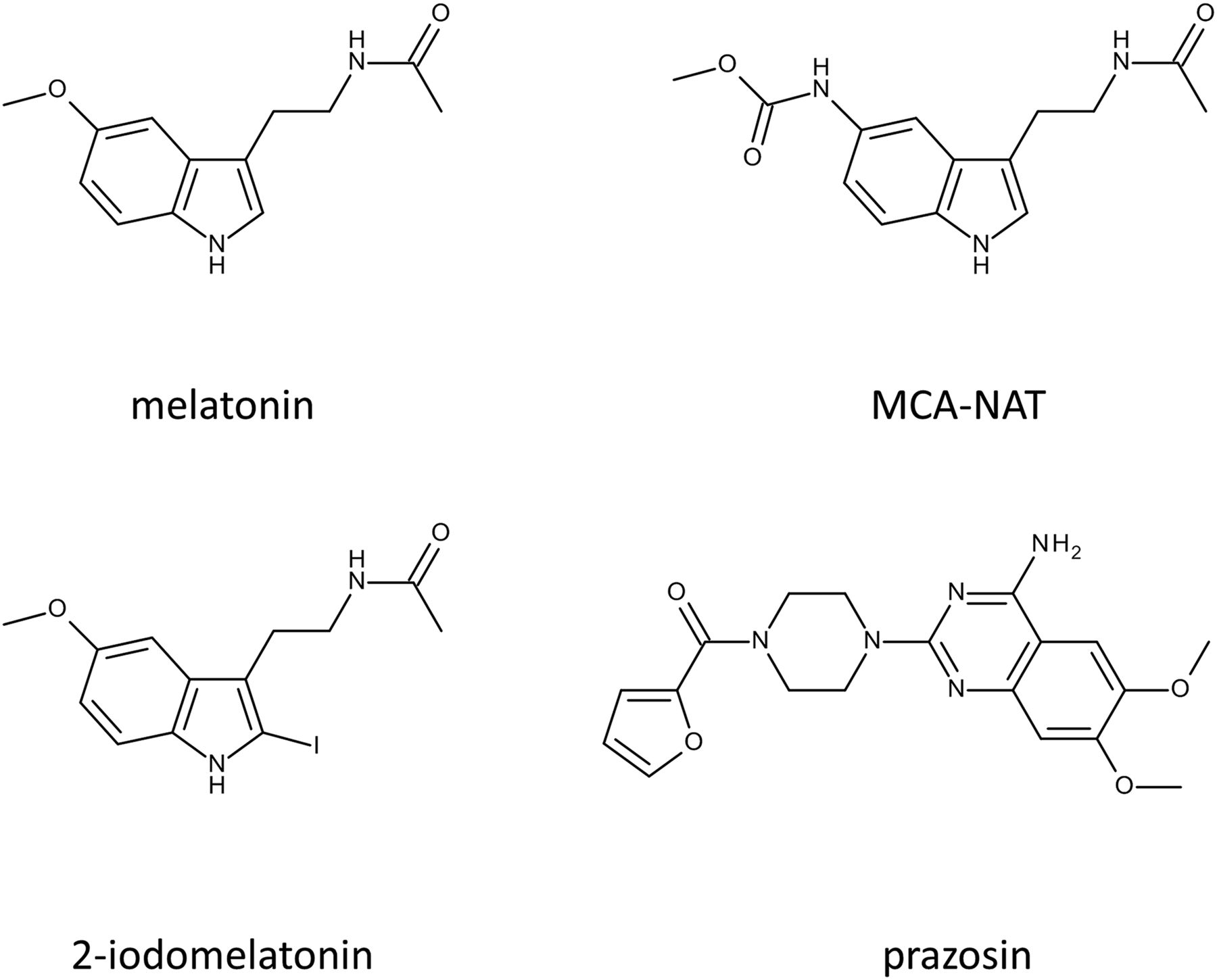
From a structural point of view, the molecules that were used to characterize the MT3 site were only loosely chemically related, particularly prazosin (see Fig. 1 for structures). We confirmed that this binding was easily observed at 4°C but not at room temperature (Paul et al., 1999; Nosjean et al., 2001). In the same articles, we also confirmed that the pharmacologic profile of MT3 was distinct from that of MT1 and MT2. The development of ultrafast binding devices made it possible to measure MT3-like binding in more standard conditions (i.e., at 20°C) (Nosjean et al., 2001), although binding was still too rapid to be detected at 37°C. These experiments were conducted with both 2-[125I]-iodomelatonin and with 2-[125I]-iodoMCA-NAT (Molinari et al., 1996).

Structures of the key molecules involved in MT3 pharmacology.
The pharmacologic profile of MT3, as determined by these later experiments, was similar to the profile that was described initially (Fig. 2); however, when interpreting the results (summarized in Fig. 2), it should be borne in mind that these studies spanned two decades, and during this time, the techniques and the approaches evolved, becoming more sensitive and more robust. Furthermore, different studies used different techniques. Finally, the biologic material used in the studies varied, with some using hamster membranes and some using human enzymes. Hamster MT1 and human MT2 melatonin receptors were the first melatonin receptors to be cloned. Their use helped clarify the nature and roles of the earlier membrane-based binding experiments. Further cloning was performed by us and by others (e.g., human, rat, mouse, sheep, hamster and other animals; see Gautier et al. (2018b) and references therein). In addition to differences in biologic materials, some studies looked at the binding of a ligand to a membrane-associated site (Fig. 2, lines 1–6), others looked at the catalytic inhibition of a pure enzyme (Fig. 2, lines 7 and 9), whereas another examined the binding of a ligand to an enzyme (Fig. 2, line 8). These differences make it difficult to compare results. As noted by Pegan et al. (2011), the resulting pharmacologic characteristics should be looked at in terms of trends and the bigger picture. Considering the differences in methods and materials in the studies, the results are quite consistent. Remarkably, the compound that showed the highest binding affinity was always the 2-iodomelatonin analog; MCA-NAT performed less efficiently than prazosin, and melatonin remained a relatively poor ligand.

The pharmacologic profiles of MT3 in different studies. The larger set of compounds was described by Molinari et al. (1996) after Pickering and Niles (1990, 1992). Assuming QR2 was the protein responsible for the MT3 binding site activity, several authors reanalyzed some molecules to classify them more fully. It is difficult to compare the studies as they used different biologic materials (hamster membranes vs. pure human QR2) and different techniques (binding vs. catalytic activity inhibition vs. isothermal titration calorimetry). Only general conclusions can be reached based on the findings overall. The compounds are colored-coded as follows: 2-iodomelatonin (2-Iodo-Mel, yellow); 6-chloromelatonin (6-Cl-Mel, light pink); [4-(4-amino-6,7-dimethoxyquinazoline-2-yl)piperazin-1-yl]-(furan-2-yl)methanone (prazosin, salmon); 5-methoxycarbonylamino-N-acetyltryptamine (MCA-NAT, blue); melatonin (Mel, deep blue); 5-hydroxytryptamin (5-OH-Trypt, pink); serotonin (Ser, green-blue). All data are reported in nanomolars for binding (lines 1–5) and in micromolars for the other parameters (lines 6–8). Compounds are shown in order from most active (far left) to least active (far right).
In summary, a membrane-associated melatonin binding site was initially described in various organs in hamsters and mice (Molinari et al., 1996; Nosjean et al., 2001; Mailliet et al., 2004), and this binding site was validated independently by several laboratories. The binding characteristics of MT3 differed from those of GPCRs, including MT1 and MT2, and its pharmacologic profile was completely different from those of other melatonin binding sites. Thus, in the early 1990s, prazosin and the undercharacterized compound MCA-NAT were the only pharmacologic tools available to investigate MT3.
MT3 Purification
Based on these data, we decided to try to purify this binding site. Our hypothesis was that it was an enzyme, since it shared characteristics with enzymes that had been characterized based on their binding capacity, like bryostatin and protein kinase C (Lewin et al., 1992) and like phorbol esters and protein kinase D (Wang, 2003). Based on our previous experience with affinity chromatography, we immobilized MCA-NAT on an affinity column; we tried two types of immobilization on two different locations of the molecule because it was unclear which part of MCA-NAT was recognized by the binding site. The structure of MCA-NAT is shown in Fig. 1. We succeeded in purifying this binding site to homogeneity from a hamster brain membrane–rich fraction after gentle solubilization with 5 mM β-octyl glucopyranoside, a mild detergent (Nosjean et al., 2000). Without a mild detergent, recovery of QR2 was poor, strongly suggesting, as proved later (Dorai et al., 2018), that at least part of QR2 was membrane-associated.
The protein was visualized as a single band on an SDS-PAGE gel and then sequenced. Analysis showed that it was an analog of human quinone reductase 2 (QR2), which was previously cloned and characterized by Talalay’s group (Zhao et al., 1997). It should be noted that QR2 can be retained on columns containing immobilized resveratrol (Buryanovskyy et al., 2004), chloroquine (Kwiek et al., 2004), or imatinib and nilotinib, two BCR-ABL inhibitors (Rix et al., 2007). These characteristics resemble those of drug metabolism enzymes, such as glutathione-S-transferases, cytochrome P450, UDP-glucuronosyltransferases, and quinone reductase 1 (Testa and Kramer, 2006, 2007, 2008). For all these enzymes, the plasticity of the catalytic site is attributed to enzyme evolution to accommodate natural and synthetic xenobiotic compounds from the environment. These enzymes eliminate specific compounds from our body and thus have broad specificity. It is widely accepted that QR2 evolved from quinone reductase 1 (Zhao et al., 1997), so QR2 could have retained these characteristics.
Characterization of MT3
Cellular Experiments.
Clearly, purifying the binding site was not sufficient to show that the MT3 binding site was the QR2 protein. We thus conducted in cellulo experiments to characterize MT3 more fully. We transfected naïve human embryonic kidney cells with QR2 and obtained a series of clones. In those clones, MT3 binding site was measurable using MT3 measurement conditions, whereas in naïve cells, it was not. All the tests performed using these cellular QR2-expressing clones showed MT3-type binding with the corresponding pharmacology (Nosjean et al., 2000). Mailliet et al. (2005) further documented this binding, reporting that additional clones that overexpressed QR2 were positive for the MT3 binding site.
In Vivo Experiments.
Next, we created QR2 knockout mice using gene deletion (Mailliet et al., 2004). Brain samples from these mice did not show MT3-type binding, but MT3 was easily measured in samples from the wild-type control animals. These knockout mice did not have any obvious phenotypes, with the complicating exception that they were reluctant to reproduce under standard laboratory conditions.
Cocrystallization.
We were curious about whether we could cocrystallize melatonin with QR2. We obtained several samples of crystal-grade enzyme and determined the three-dimensional structure of the melatonin inside the enzyme’s catalytic site (Calamini et al., 2008). We also resolved cocrystals with a series of QR2-related compounds, including resveratrol [previously cocrystallized with QR2 by Buryanovskyy et al. (2004)] and the reference MT3 compound, MCA-NAT, and its derivative, 2-iodoMCA-NAT (Pegan et al., 2011), and, of course, prazosin, which was also cocrystalized in hQR2 (Pegan et al., 2011).
Challenging Attempts to Measure the Binding of Melatonin to Pure QR2 Enzyme
Using the purified QR2, we tested several technical solutions to measure the binding of melatonin at QR2 to compare it with the inhibitory potency of melatonin at QR2 catalytic activity. Several data sets were compiled from isothermal titration calorimetry (ITC) experiments, which measure the heat dispersion when a compound binds to a protein. Binding of melatonin to QR2 was measured directly using ITC (Calamini et al., 2008), and the Kd was 1 µM; in contrast, the inhibition capacity is in the 10–130 µM range, depending on the system used to measure the inhibition (Calamini et al., 2008; Ferry et al., 2010; Pegan et al., 2011). These data showed that melatonin, 2-iodomelatonin, and resveratrol all had high affinities for QR2. Notably, resveratrol was used as a reference for QR2 binding based on the observations of Buryanovskyy et al. (2004). The order of potency of those three compounds was as expected: melatonin and 2-iodomelatonin bound well to QR2, and the orders of magnitude of the binding were in accordance with the original characterization of the MT3 binding site (see Fig. 2 for details). We further studied the compound and enzyme relationship using mass spectrometry in native mode and confirmed the binding of melatonin and resveratrol to QR2 (Antoine et al., 2012).
Enzyme binding experiments require great care, as illustrated by the binding of phorbol esters to protein kinase C (Chauhan et al., 1989; Dimitrijevic et al., 1995). Our laboratory tried to immobilize QR2 by a His-tag tail to nickel-bearing particles and then use this as a standard binding site for either 2-iodo-melatonin or 2-iodoMCA-NAT. Unfortunately, we were unable to obtain reliable results (S. Coumailleau, G. Ferry, O. Nosjean, J. A. Boutin, unpublished data).
Use of Indirect Pharmacologic Evidence to Suggest Implication of MT3
Melatonin, resveratrol, prazosin, and MCA-NAT can all bind to more than one cellular target. This is widely accepted for resveratrol (Yang et al., 2015) and melatonin (Reiter et al., 2016) but may be less clear for prazosin and MCA-NAT. Prazosin is a specific adrenoceptor ligand that has been the subject of several reviews, including one by Brogden et al. (1977). Prazosin has two off-targets—namely, MT3 (Molinari et al., 1996; Paul et al., 1999) and 5HT2A (IC50 0.7 nM) (Andorn et al., 1984). The affinity of prazosin for the MT3 binding site ranges from 7.4 to 10.2 nM, and prazosin binds to the α1-adrenoceptor at nanomolar concentrations, particularly to the α1D subtype (pKi 10.2) (Ford et al., 1997). Indeed, prazosin is the reference ligand for the α1D adrenoceptor.
MCA-NAT is described as binding specifically to the MT3 binding site, but this is not completely accurate. Whereas the compound undoubtedly has a preferred affinity for this binding site, it is also a good agonist for both melatonin receptors, with affinities of 1 µM and lower (Dubocovich et al., 1997; Paul et al., 1999; Nosjean et al., 2001; Vincent et al., 2010), and MCA-NAT cocrystalizes with QR2 (Pegan et al., 2011). Furthermore, Crooke et al. (2011) showed that MCA-NAT downregulates the CAXII and CAII carbonic anhydrases and noted that these enzymes are molecular targets of MCA-NAT. MCA-NAT also downregulates the β2 adrenoceptor and upregulates the α2A adrenoceptor (Crooke et al., 2011).
Some of the best known experiments with MCA-NAT are those that address intraocular pressure (IOP) in various animal species and models (Pintor et al., 2001, 2003; Serle et al., 2004; Alarma-Estrany et al., 2009, 2011; Andres-Guerrero et al., 2009, 2011; Crooke et al., 2011, 2012, 2013; Dortch-Carnes and Tosini, 2013; Huete-Toral et al., 2015; Martinez-Aguila et al., 2016; Zaryanova et al., 2017). These studies consistently showed that MCA-NAT treatment decreased IOP. They also showed that QR2 was not involved in this effect and that the effect was sensitive to the melatonin receptor antagonists luzindole and/or 4P-PDOT (Pintor et al., 2001; Dortch-Carnes and Tosini, 2013; Martinez-Aguila et al., 2016). Incidentally, luzindole is a poor ligand for MT3 (Molinari et al., 1996). An elegant experiment used anti-QR2 siRNA to knock down QR2 expression; in these conditions, the action of MCA-NAT was maintained (Alarma-Estrany et al., 2009). This could be interpreted two ways. First, one could conclude that since there was no QR2, the target of MCA-NAT is not QR2 (thus MT3 is not QR2). Second, one could conclude that since there was no QR2 or antagonism owing to luzindole and/or 4P-PDOT (both of which are reference antagonists for melatonin receptors), and since MCA-NAT is a mild agonist of melatonin receptors, the observed effects were due not to MT3/QR2 but rather to MT1 and/or MT2.
The conclusions of such pharmacologic studies implying antagonists or inhibitors are always limited in that there is some question as to the specificity of those pharmacologic agents.
Membrane Association: The Crux of the Question of Whether MT3 is QR2
It is important to keep in mind that MT3 was first identified as a membrane-bound melatonin binding site using 2-iodomelatonin binding (Duncan et al., 1988) and that its pharmacologic profile differed from those of the other melatonin receptors (Dubocovich, 1995). In contrast, QR2 has been reported to be a cytosolic enzyme, including by our group. Nevertheless, the purification of QR2 from naturally expressing QR2 cells always necessitates the use of a mild detergent (β-octyl glucopyranoside), suggesting that at least part of QR2 is associated with cell membranes (see also later). This difference (i.e., the fact that MT3 was originally described as being membrane-associated and that QR2 was nonmembrane-associate) is the basis of the controversy regarding whether MT3 and QR2 are the same entity; however, the indirect evidence seems to indicate that they are the same. Western immunoblotting analysis of membrane proteins from QR2-overexpressing cells using an anti-QR2 antibody showed a clear signal for membrane-associated QR2. It is possible that this was an artifact owing to enzyme overexpression (N. Moulharat, G. Ferry, and J. A. Boutin, unpublished data). Furthermore, the use of a mild detergent is determinant in obtaining a good recovery of QR2 prior purification. An article from the Wu group (Dorai et al., 2018) reported “significant localization of NQO2 (a.k.a. QR2) in the detergent-insoluble membrane fraction.” Those experiments clearly show that at least some QR2 proteins are associated with membranes. Taken together, these data indicate that in some conditions, QR2 is observed in membrane-rich fractions, even in fractions from cells that do not overexpress QR2. This merits further study.
Toward its N terminus, QR2 has a cryptic myristoylation site (Gly34). Myristoylation is an N-terminal glycine-based post-translational modification that is mediated by the N-myristoyltransferase enzyme (Boutin, 1997). This enzyme recognizes a short sequence in the N-terminal region of target proteins (provided the N-terminal amino acid is glycine) and transfers a myristate moiety from myristoyl-coenzyme A to the glycine residue. Many proteins can be modified in this manner, ranging from the src oncogene to the gag structural protein of RNA viruses (Boutin, 1997). Myristoylation allows a protein to become membrane-associated and to phosphorylate (src) nearby proteins or direct the formation of viral particles (gag). While studying another protein, we found that cryptic myristoylation sites could be digested by caspase proteolysis activity, creating a free N-terminal glycine that can be myristoylated, and then the protein could associate with membranes (Degli Esposti et al., 2003). We were not able to express either a Gly-to-Ala QR2 mutant or a QR2 protein that was truncated from the N terminus of the glycine in the cryptic site (Gly34) using an Escherichia coli expression system (S. P. Guenin and J. A. Boutin, unpublished data).
Another possibility is that QR2 could dimerize with a membrane-associated protein. Such dimerization has been reported for QR2 and p53 (Gong et al., 2007), although the report was later withdrawn. Our laboratory was unable to coimmunoprecipitate QR2 using anti-p53 antibody from cells that naturally express QR2. Others have suggested the possibility of heterodimerization with caveolin (Dorai et al., 2018).
In conclusion, it seems extremely reasonable to suggest that QR2 could be associated with plasma membranes in vivo and that the QR2 protein is the same as the MT3 melatonin binding site.
Perspectives
If the enzyme QR2 is in fact MT3, this presents new possibilities in terms of approaches to investigating melatonin functionality [see discussion in Boutin (2015)]. It may also provide insights into the mechanisms underlying the therapeutic effects of melatonin on many pathologies at pharmacologic doses (micromolar and higher). Slominski et al. (2012) noted that melatonin receptors are attractive targets for the “immunomodulation, regulation of endocrine, reproductive and cardiovascular functions, modulation of skin pigmentation, hair growth, cancerogenesis [sic], and aging” [see Boutin (2015) for 200 additional examples]. We and others have demonstrated that QR2 can transform quinones, particularly ortho-quinones, such as dopamine-quinone (Cassagnes et al., 2015), into their unstable quinol counterparts (Reybier et al., 2011). In the presence of oxygen, quinols oxidize back into quinones, completing a futile cycle that generates massive bursts of reactive oxygen species. This cycle can be observed in cells in which QR2 has not been overexpressed (Cassagnes et al., 2015), including in neurons (Cassagnes et al., 2018). Thus, depending on the circumstances (which are not well understood), the enzyme can function in toxification rather than as a detoxification enzyme. Notably, in QR2 knockout mice, menadione is far less toxic than in wild-type animals (Long et al., 2002). Our laboratory has independently confirmed these intriguing results (P. Delagrange and J. A. Boutin, unpublished data). More data are needed to show clearly that there is a relationship between QR2 and some pathologies, but this body of evidence may explain why melatonin can be active at pharmacologic concentrations as a QR2 inhibitor.
Conclusions
The available data indicate that QR2 is indeed MT3: with one exception, the purification, expression, pharmacologic, and structural data show that the binding site and the enzyme are the same, and experiments in knockout mice also support this conclusion. The recent finding that a portion of QR2 is associated with membranes further supports the conclusion that QR2 is MT3. Only indirect evidence from pharmacologic experiments suggests otherwise; notably, the compounds used in those studies were not specific to MT3 and thus might incorrectly implicate MT3 while interacting with other protein targets. Specifically, MCA-NAT may act as a melatonin receptor agonist at micromolar concentrations.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Ferry, Boutin.
Footnotes
- Received August 28, 2018.
- Accepted October 26, 2018.
Abbreviations
- GPCR
- G-protein–coupled receptors
- ITC
- isothermal titration calorimetry
- MCA-NAT
- 5-methoxycarbonylamino-N-acetyltryptamine or GR135531
- MT3
- the third melatonin binding site (formerly known as ML2)
- QR2
- quinone reductase 2, also called NQO2, N-ribosyldihydronicotinamide:quinone reductase 2
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- History of MT3 Binding Site
- MT3 Purification
- Characterization of MT3
- Challenging Attempts to Measure the Binding of Melatonin to Pure QR2 Enzyme
- Use of Indirect Pharmacologic Evidence to Suggest Implication of MT3
- Membrane Association: The Crux of the Question of Whether MT3 is QR2
- Perspectives
- Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters