


Abstract
A combination of buprenorphine (BUP) and samidorphan (SAM) at a 1:1 (mg/mg) fixed-ratio dose is being investigated as an adjunctive treatment of major depressive disorder (BUP/SAM, ALKS 5461). Both [3H]BUP and [3H]SAM bound to the μ-, κ-, and δ-opioid receptors (MOR, KOR, and DOR, respectively) with Kd values of 3 nM or less. [3H]BUP dissociated from the MOR more slowly than [3H]SAM did. In the [35S]GTPγS assay, BUP was a partial agonist at the MOR, KOR, and DOR. SAM was an antagonist at the MOR and a partial agonist at the KOR and DOR. The pharmacology of the combination of SAM and BUP was characterized at ratios like the molar ratios of both compounds at steady state in humans. In all assessments, SAM reduced the efficacy of BUP at the MOR without altering its potency. At the KOR, SAM had no significant effect on the activity of BUP. In bioluminescent resonance energy transfer assays, SAM, naltrexone, and naloxone were partial agonists when the MOR was coupled to the GαoB and Gαz, and were antagonists when coupled to Gαi. At the KOR, SAM was a partial agonist activating GαoA and GαoB and a full agonist in stimulating Gαz. SAM inhibited BUP’s recruitment of β-arrestin to the MOR, suggesting an attenuation of BUP’s efficacy in activating G proteins correlated with an inhibition of β-arrestin recruitment. The collective data suggest that SAM attenuates the efficacy of BUP under all conditions tested at the MOR and DOR but had little effect on BUP activity at the KOR.
Introduction
BUP/SAM (ALKS 5461) (Fig. 1) consists of the partial μ-opioid agonist buprenorphine (BUP) and the μ-opioid antagonist samidorphan (SAM) at a 1:1 (mg/mg) dose ratio. The combination is being studied as adjunctive therapy in patients having an inadequate response to antidepressants (Ehrich et al., 2015; Fava et al., 2016) and represents a potential treatment of major depressive disorder (MDD) working through opioid receptor modulation.

Structures of SAM and BUP.
Numerous lines of evidence suggest BUP’s potential in treating depression. In humans, BUP has been shown to have antidepressive effects in treatment-resistant patients (Bodkin et al., 1995; Nyhuis et al., 2008; Kosten, 2016). Preclinical assessments support a role for the μ-, κ-, and δ-opioid receptors (MOR, KOR, and DOR, respectively) in the treatment of depression (Lutz and Kieffer, 2013) and a role for both the MOR and KOR has been postulated for BUP in producing antidepressive and antianxiolytic effects (Falcon et al., 2015, 2016; Robinson et al., 2017; Almatroudi et al., 2018). SAM was synthesized as a naltrexone derivative containing a 3-carboxamido group and was shown to have high affinity for the MOR (Wentland et al., 2005) and to act as an antagonist at the MOR and a partial agonist at the KOR and DOR (Wentland et al., 2009). In preclinical studies, appropriate titration of SAM attenuates the abuse potential of BUP, at the same time having no effect on the antidepressant efficacy (Smith et al., 2017).
Despite BUP’s clinical use, relatively little is known about the receptor binding and in vitro pharmacological properties of BUP at the MOR, KOR, and DOR and the nociceptin/orphanin FQ (NOP) receptor. The few previous [3H]BUP binding experiments were performed before the cloning of the multiple opioid receptors and used rodent brain and spinal cord membranes (Villiger and Taylor, 1981, 1982; Villiger, 1984; Boas and Villiger, 1985). [3H]BUP binding to rat spinal cord membranes gave a curvilinear Scatchard plot, indicative of multiple binding sites (Villiger and Taylor, 1982).
Activation of the MOR results in signaling through Gαi/o proteins to inhibit adenylyl cyclase activity, activate K+ channels, inhibit Ca2+ channels, and activate mitogen-activated protein kinases (Law et al., 2000; Lamberts et al., 2018). BUP is a partial agonist at the MOR (Huang et al., 2001). On the basis of animal and human studies, some investigators characterize BUP as a KOR antagonist (Leander, 1987), although previous [35S]GTPγS binding studies have reported it is a partial agonist at the KOR (Huang et al., 2001; Wentland et al., 2009). BUP was a DOR antagonist in behavioral studies and in [35S]GTPγS binding studies using membranes from the NG108-15 cell line (Negus et al., 2002). There is general agreement that BUP is a partial agonist at the NOP receptor but it is less potent at the NOP receptor than at the opioid receptors (Huang et al., 2001; Cami-Kobeci et al., 2011; Khroyan et al., 2015).
Until recently, it was not possible to study the signaling of an opioid receptor interacting with a specific G protein subunit. A bioluminescent resonance energy transfer (BRET) assay with the MOR expressed with a specific Gα protein, endomorphin-1, a μ-selective opioid agonist, showed maximal efficacy when the MOR was signaling through GαoA and GαoB but only partial agonist activity when the MOR was signaling through Gαi1, Gαi2, Gαi3, or Gαz (Masuho et al., 2015). Likewise, at the KOR, the κ-endogenous peptide dynorphin A produced maximal efficacy when the KOR signaled through GαoA and GαoB and had partial agonist activity when the KOR signaled through Gαi1, Gαi2, Gαi3, or Gαz. Enkephalin produced a similar response at the DOR (Masuho et al., 2015). These data suggest opioids signal promiscuously but with varying activity through the multiple G proteins.
The recruitment of β-arrestin by opioids has been associated with the deleterious effects associated with opioid use (Schmid et al., 2017). Hence, compounds that bias away from β-arrestin recruitment have garnered great interest. A number of compounds such as BUP (McPherson et al., 2010; Chen et al., 2013; Grinnell et al., 2016), oliceridine (TRV130) (Chen et al., 2013), mitragynine and 7-hydroxymitragynine (Kruegel et al., 2016), and PZM21 (Manglik et al., 2016), produce little or no β-arrestin recruitment to the MOR. However, these molecules are also partial agonists in activating Gαi/o signaling mediated by the MOR.
The goal of the current study was to understand the in vitro pharmacological properties of BUP and SAM alone as well as in combination at molar ratios like those observed in humans upon administration of BUP/SAM combination. Given its probable mechanistic role in MDD treatment, the activity of MOR and KOR in combination was characterized more extensively. Additionally, the diversity of techniques employed allowed an assessment of comparability of various assay systems.
Materials and Methods
Compounds and Cells.
SAM (3-carboxyamido-4-hydroxy naltrexone) was synthesized as previously described (Wentland et al., 2005). BUP-HCl was obtained from Siegfried USA (Pennsville, NJ). DAMGO, U50,488-HCl hydrate, and SNC-80 were obtained from Sigma-Aldrich Corp. (St. Louis, MO). [Tyrosyl-3,5-3H(N)] DAMGO (47.1 Ci/mmol), [phenyl-3,4-3H]U69,593 (44.6 Ci/mmol), [leucyl-3,4,5-3H]nociception (123.6 Ci/mmol), and [35S]GTPγS were obtained from PerkinElmer (Waltham, MA). [5,7-3H] Naltrindole (30 Ci/mmol) and [ring-3H]BUP-HCl (20 Ci/mmol) were obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO). [3H]SAM (20 Ci/mmol) was provided by Moravek Biochemicals (Brea, CA).
Chinese hamster ovary (CHO) cells stably expressing the human MOR, KOR, or DOR (hMOR-CHO, hKOR-CHO, or hDOR-CHO) and CHO cells expressing the NOP receptor were used in this study. hMOR-CHO cells were provided by Dr. George Uhl (NIDA Intramural Program, Baltimore, MD) and hKOR-CHO cells were provided by Dr. Lee-Yuan Liu-Chen (Temple University, Philadelphia, PA). hDOR-CHO and CHO cells stably expressing the human NOP receptor were obtained from Dr. Larry Toll (Florida Atlantic University, Boca Raton, FL).
Membrane Preparations.
hMOR-CHO, hKOR-CHO, or hDOR-CHO cells and CHO cells stably expressing the NOP receptor were cultured in 100-mm dishes in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum and penicillin−streptomycin (10,000 IU/ml) at 37°C in a 10% CO2 atmosphere. When the cells were at confluence, they were gently scraped from the 100-mm dishes and added to a 50-ml centrifuge tube. The cells were centrifuged at 1000g for 10 minutes at 4°C. Cell pellets prepared for radioligand binding assays were resuspended in 50 mM Tris-HCl, pH 7.4. Resuspended cell pellets from all centrifuge tubes were combined and homogenized using a Polytron cell homogenizer. The homogenized preparation was centrifuged at 18,000g for 30 minutes at 4°C. The pellet was resuspended in 40 ml of cold 50 mM Tris-HCl, pH 7.4, and the centrifugation step was repeated. The final membrane pellet was resuspended in 50 mM Tris-HCl, pH 7.4, at a protein concentration between 3 and 5 mg/ml. These membranes were used in the radioligand receptor binding assays. Cell membranes prepared for [35S]GTPγS assays were resuspended in phosphate-buffered saline buffer containing 0.04% EDTA and centrifuged at 1000g for 10 minutes at 4°C. The supernatant was removed and the cell pellet resuspended and homogenized in 50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4 (membrane buffer). The homogenized preparation was centrifuged as described above and the final membrane pellet resuspended in 50 mM Tris-HCl, 3 mM MgCl2, 100 mM NaCl, 0.2 mM EGTA, pH 7.4 (assay buffer), at a membrane protein concentration between 3 and 5 mg/ml. Membrane protein concentrations were determined with bovine serum albumin as the control as previously described (Bradford, 1976). Membranes were stored at −80°C until use.
Radioligand Competition Binding Assays.
To determine the binding affinities (Ki values) of BUP and SAM for MOR, KOR, DOR, and NOP receptors, 12 different concentrations of each compound were incubated with a specific radioligand and CHO membranes expressing a single type of receptor in 50 mM Tris-HCl, pH 7.4, at a final volume of 1 ml. The radioligand concentrations used were 0.25 nM [3H]DAMGO, 0.4 nM [3H]U69,593, 0.2 nM [3H]naltrindole, and 0.1 nM [3H]nociceptin, for measuring MOR, KOR, DOR, and NOP receptor binding, respectively. Nonspecific binding was measured using 10 μM naloxone in experiments with the μ-selective peptide [3H]DAMGO and the κ-selective alkaloid [3H]U69,593, and 100 μM naloxone in experiments with the δ antagonist [3H]naltrindole. Unlabeled nociceptin, at 1 μM, was used to measure nonspecific binding in assays with [3H]nociceptin. Binding was initiated with the addition of CHO membranes (50 μg of membrane protein for MOR, 25 μg for KOR, 20 μg for DOR, and 2 μg for NOP receptor). Assays were incubated for 60 minutes except assays with [3H]naltrindole, which required a 3-hour incubation. All assays were performed at 25°C and terminated by rapid vacuum filtration through Whatman #32 glass fiber filters using a Brandel cell harvester (Gaithersburg, MD) followed by three ice-cold washes with 50 mM Tris-HCl, pH 7.4. Filters were presoaked for 60 minutes in 0.25% polyethyleneimine (PEI) solution to reduce free radioligand binding to the filter. Binding assays with [3H]DAMGO did not require PEI-soaked filters. Samples were collected and counted in 2 ml of Ecoscint A scintillation fluid (National Diagnostics, Atlanta, GA) for 2 minutes each, using an LS 6500 scintillation counter (Beckman Coulter Inc., Fullerton, CA).
To determine the affinity of BUP, SAM and other opioid full and partial agonists and antagonists under high- and low-affinity binding conditions at the MOR, binding experiments were performed as previously described with some modifications (Emmerson et al., 1996). High-affinity binding was measured in 50 mM Tris-HCl buffer, pH 7.4. Low-affinity binding was measured in 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, and 50 μM GDP (GP buffer) as previously described (Emmerson et al., 1996). To measure high-affinity binding, we used the μ-selective agonist [3H]DAMGO (0.25 nM) instead of [3H]sufentanil, which had been used previously (Emmerson et al., 1996). To measure low-affinity binding, [3H]naloxone (1.0 nM) was used instead of [3H]naltrexone (Emmerson et al., 1996). In a final volume of 1 ml, hMOR-CHO membranes, 50 μg of protein, were incubated either in 50 mM Tris-HCl, pH 7.4, or GP buffer containing eight different concentrations of the compound and either [3H]DAMGO to measure high-affinity binding or [3H]naloxone to measure low-affinity binding. Unlabeled naloxone at a final concentration of 10 μM was used to measure nonspecific binding. Membranes were added last to the incubation tube. After a 60-minute incubation at 25°C, membranes were filtered through Whatman #32 glass fiber filters, followed by washing with cold 50 mM Tris-HCl, pH 7.4, and counting of the filters as described above.
For competition binding experiments, the specific binding of the radioligand in the absence of other competing compounds was set at 100%. The percent of control binding was calculated for the increasing concentrations of the competing compounds. A nonlinear regression curve was fit to the data using SigmaPlot. The IC50 values, the concentration of the opioid needed to inhibit 50% of the control binding, were calculated from the curves. Ki values were calculated as described previously (Cheng and Prusoff, 1973). Data are reported as the mean Ki value ± S.E.M. from three separate experiments performed in triplicate.
Radioligand Saturation Binding Assays and Competition Binding Experiments.
We characterized the binding affinity (Kd) and maximum binding (Bmax) of [3H]BUP and [3H]SAM under high- and low-affinity binding conditions to hMOR-, hKOR-, and hDOR-CHO membranes. High-affinity binding was measured in 50 mM Tris-HCl buffer, pH 7.4. Low-affinity binding was measured in GP buffer as described above. Eight concentrations of either [3H]BUP or [3H]SAM were incubated with CHO membranes expressing either the MOR, KOR, or DOR in 50 mM Tris-HCl, pH 7.4 or GP buffer at a final volume of 1 ml. Nonspecific binding was measured with 10 μM naloxone. CHO membranes were added last. The amount of membrane used was chosen to maximize the specific signal at the same time keeping specific binding below 10% of total binding. Saturation binding assays with [3H]BUP used 100 μg hMOR-CHO, 15 μg hKOR-CHO, and 10 μg hDOR-CHO membrane protein; those with [3H]SAM used 20 μg hMOR-CHO, 25 μg hKOR-CHO, and 20 μg hDOR-CHO membrane protein. Binding was terminated by rapid vacuum filtration through PEI-soaked glass fiber filters after a 90-minute incubation for [3H]BUP and after 60-minute incubation at 25°C for [3H]SAM. Samples were washed and counted as described above.
SAM, BUP, naltrexone, and naloxone were tested to determine the Ki values for these compounds in inhibiting [3H]BUP and [3H]SAM binding to the MOR in Tris-HCl and GP buffers. Membrane protein, 100 μg, from hMOR-CHO cells was incubated with 0.4 nM [3H]BUP for 90 minutes or 0.05 nM [3H]SAM for 60 minutes at 25°C in a final volume of 1 ml in either Tris-HCl, pH 7.4, or GP buffers. Nonspecific binding was measured by the inclusion of 10 μM naloxone. The compounds were tested at 12 different concentrations and membranes were added last to the sample. A nonlinear regression curve was fit to the data using SigmaPlot. The IC50 values were calculated from the curves. The Kd values for [3H]BUP and [3H]SAM in both buffers were used to calculate the Ki values. Ki values were calculated as described previously (Cheng and Prusoff, 1973). Data are reported as the mean Ki value ±S.E.M. from three separate experiments performed in triplicate.
Radioligand Association and Dissociation Assays.
We investigated the kinetic binding properties of [3H]BUP and [3H]SAM at MOR, KOR, and DOR under high- and low-affinity binding conditions. Association assays were performed by incubating the radioligands with hMOR-CHO, hKOR-CHO, and hDOR-CHO cell membranes for various times in 50 mM Tris-HCl, pH 7.4 or GP buffer. Nonspecific binding was measured at each time point by addition of 10 μM naloxone. Dissociation assays were performed by incubating the radioligands with membranes (90 minutes for [3H]BUP and 60 minutes for [3H]SAM) to reach equilibrium at 25°C, and then adding unlabeled BUP (10 μM) or SAM (1 μM) at different times to displace the bound radioligand. Concentrations of [3H]BUP and [3H]SAM near their respective Kd values under high- and low-affinity binding conditions were used in both the association and dissociation assays. [3H]BUP concentrations were 0.4, 0.2, and 1.5 nM for high-affinity binding assays, and 0.4, 0.1, and 0.5 nM for low-affinity binding assays at MOR, KOR, and DOR, respectively. [3H]SAM concentrations were 0.05, 0.2, and 1.0 nM for high-affinity binding assays, and 0.05, 2.0, and 1.5 nM for low-affinity binding assays at MOR, KOR, and DOR, respectively. Membrane protein amounts used for association and dissociation assays at each receptor under high- and low-affinity binding conditions were the same as those used for the saturation binding assays. All assays were terminated by rapid vacuum filtration through PEI-soaked filters and filters were washed and counted as described above.
Association binding data were analyzed with SigmaPlot, using a nonlinear regression through the origin. The association T1/2 value, the time required to reach 50% of maximal binding, was calculated from the curve. Dissociation binding data were expressed as percent of control binding. Control binding is the specific binding measured at equilibrium (90 minutes for [3H]BUP and 60 minutes for [3H]SAM) and was set at 100%. Nonlinear regression curves were fit to the data using SigmaPlot. The time required for dissociation of 50% of the control binding (dissociation T1/2) was determined from the curve. Kon and Koff rates and Kd values were calculated using GraphPad Prism (La Jolla, CA). The data are reported as mean ± S.E.M. from three separate experiments, performed in triplicate.
[35S]GTPγS Binding Assays.
Agonist and antagonist properties of BUP and SAM were investigated using the [35S]GTPγS binding assay to measure G protein activation in response to ligand-receptor binding. Various concentrations of either BUP or SAM were incubated with hMOR-, hKOR- or hDOR-CHO membranes for 60 minutes at 30°C in assay buffer with 3 μM GDP and 0.08 nM [35S]GTPγS in a final volume of 0.5 ml. Unlabeled GTPγS, at 10 μM, was used to measure nonspecific binding. As controls, maximal stimulation was measured with 10 μM concentrations of DAMGO, U50,488, or SNC80 for the MOR, KOR, and DOR, respectively, and was set at 100%. The agonist effects of 1:1, 1:5, and 1:50 molar concentrations of BUP to SAM in [35S]GTPγS binding assays at MOR and KOR were also determined. The data are expressed as percent stimulation of [35S]GTPγS binding. Nonlinear regression curves were fit to the data using SigmaPlot software. Maximal stimulation (Emax) values and the concentrations producing 50% maximal stimulation (EC50 values) were calculated from the curves. Data are expressed as mean ± S.E.M. from three separate experiments, performed in triplicate.
Antagonist activity of BUP or SAM was determined in the presence of a concentration of full agonist that produced approximately 80% of a maximal response alone. The concentrations of the full agonists were, 200 nM DAMGO (μ), 30 nM U50,488 (κ), or 10 nM SNC-80 (δ), and the maximum agonist stimulation was set at 100%. Maximum antagonism was determined using a pure antagonist in the presence of an agonist and represents the lower limit, 0% stimulation. Naloxone, at 10 μM, was used for experiments at MOR and DOR, whereas 1 μM nor-binaltorphimine was used for experiments at the KOR. All assays were initiated by the final addition of CHO membranes and incubated for 60 minutes in a 30°C water bath. The assays were terminated using rapid vacuum filtration through glass fiber filters. The samples were washed three times with ice-cold 50 mM Tris-HCl, pH 7.4, collected, and counted as described above. Imax values, the maximal inhibition of the agonist stimulation, and IC50 values, the concentration of compound producing 50% of the maximal inhibition, were calculated from nonlinear regression curves generated in SigmaPlot. Data are expressed as mean ± S.E.M. from three experiments performed in triplicate.
Bioluminescence Resonance Energy Transfer Assays.
Human embryonic kidney 293 (HEK293) cells and BRET biosensor constructs (Namkung et al., 2016) were provided by Domain Therapeutics, Inc. (Montreal, Canada). Cells were grown in DMEM, supplemented with 10% fetal bovine serum, 1X penicillin and streptomycin, and were maintained at 37°C in a humidified incubator containing 5% CO2. All cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA). Cells were detached, counted, and resuspended into cell culture medium. Transfection with receptor and biosensor plasmids was performed using 3 μg/ml of PEI as the transfection reagent, and 1.8 × 106 cells in T-75 cell culture flask. Serial dilutions of test compounds were prepared in 100% dimethyl sulfoxide using the HP D300 digital dispenser, and 384-well low-volume plate (white opaque; PerkinElmer). Forty-eight hours after transfection, cells were washed once with phosphate buffered saline, removed with trypsin, centrifuged at 1000g, and the pellet was resuspended in Tyrode-HEPES buffer (Boston BioProducts, Ashland, MA). Cells (10,000 cells per well) were dispensed into premade compound plates containing an opioid using the automated BioTek Multi-Flo reagent dispenser, and incubated at room temperature for 50 minutes. Coelenterazine (NanoLight Technologies, Pinetop, AZ) was subsequently added at a final concentration of 2 μM. Cells were incubated for an additional 10 minutes at room temperature, and BRET readings were captured using the Envision plate reader (filters: 400/70 nm, 515/20 nm; PerkinElmer). BRET signals were determined by calculating the ratio of light emitted by GFP-acceptor (515 nm) over light emitted by luciferase-donor (400 nm). For each test compound/biosensor pair, the test compound-induced BRET signals were normalized to the BRET signal obtained from DAMGO, U50,488, and SNC-80 for the MOR, KOR, and DOR, respectively. Maximal and minimal BRET signals were defined by 20 μM DAMGO/20 μM U50,488/20 μM SNC-80, or cells in the absence of agonists, respectively.
Results
Radioligand Competition Binding Experiments.
Using membranes from hMOR-, hKOR-, and hDOR-CHO cells, competition binding experiments showed that BUP had the highest affinity for the human KOR with a Ki value of 0.23 ± 0.0067 nM, with 2- and 10-fold lower affinity for the MOR and DOR, respectively (Table 1). SAM had the highest affinity for the MOR with a Ki value of 0.052 ± 0.0044 nM, 8-fold higher than BUP’s affinity for the MOR. SAM had essentially the same affinity as BUP for the KOR and DOR. BUP bound to the NOP receptor with a Ki value that was more than 400-fold greater than observed at the KOR and MOR, and SAM did not inhibit [3H]nociceptin binding at concentrations up to 10 μM.
Ki values for BUP and SAM in inhibiting binding to the MOR, KOR, DOR, and NOP receptor
Membranes from CHO cells expressing one type of human opioid receptor or the nociceptin/FQ receptor were incubated with 12 different concentrations of BUP or SAM. IC50 values were determined and Ki values were calculated. Data are the mean Ki value ± S.E.M. from three independent experiments performed in triplicate.
Ki values obtained for BUP and SAM were next determined for the MOR under conditions favoring high- and low-affinity binding states. High-affinity binding was performed in 50 mM Tris-HCl, pH 7.4, buffer with [3H]DAMGO as the radioligand. Low-affinity binding was measured in GP buffer with [3H]naloxone as the radioligand. BUP and SAM were compared with a series of full agonists, partial agonists, and antagonists (Table 2). The ratio of the Ki value in the low-affinity receptor state divided by the Ki value obtained for the compound with the receptor in the high-affinity state was indicative of the preference for one binding condition over the other. Full MOR agonists demonstrated up to a 530-fold preference for the high-affinity binding state over the low-affinity state, and a rank order of preference of morphine > methadone > fentanyl ≥ DAMGO. These findings agree with previous results (Emmerson et al., 1996). Partial agonists also demonstrated greater affinity for the MOR when the binding experiment was performed with the high-affinity 50 mM Tris-HCl, pH 7.4, buffer instead of the low-affinity GP buffer, though the shifts were smaller than with full agonists (Table 2). The exception was BUP, which had a 2.5-fold higher affinity for the MOR in the GP buffer. Indeed, the pattern of activity with BUP was like the MOR antagonists, which all showed less than a 2-fold shift in binding affinity between the assay conditions. SAM had a 2-fold higher affinity for the MOR under low-affinity binding condition. Hence, both SAM and BUP showed little change in Ki values upon shifting from low- to high-affinity binding conditions. The fact that BUP had a higher affinity for the MOR in the low-affinity binding condition suggests that this unique property may contribute to physiologic effects observed with BUP.
Affinities of full and partial opioid agonists and antagonists for binding to the MOR under high- and low-affinity binding conditions
Klow/Khigh is the Ki value obtained under low-affinity binding conditions in GP buffer divided by the Ki value obtained under high-affinity Tris-HCl buffer conditions. Data are the mean Ki values ± S.E.M. from three separate experiments performed in triplicate.
Association and Dissociation Rates for [3H]BUP and [3H]SAM at the Multiple Opioid Receptors.
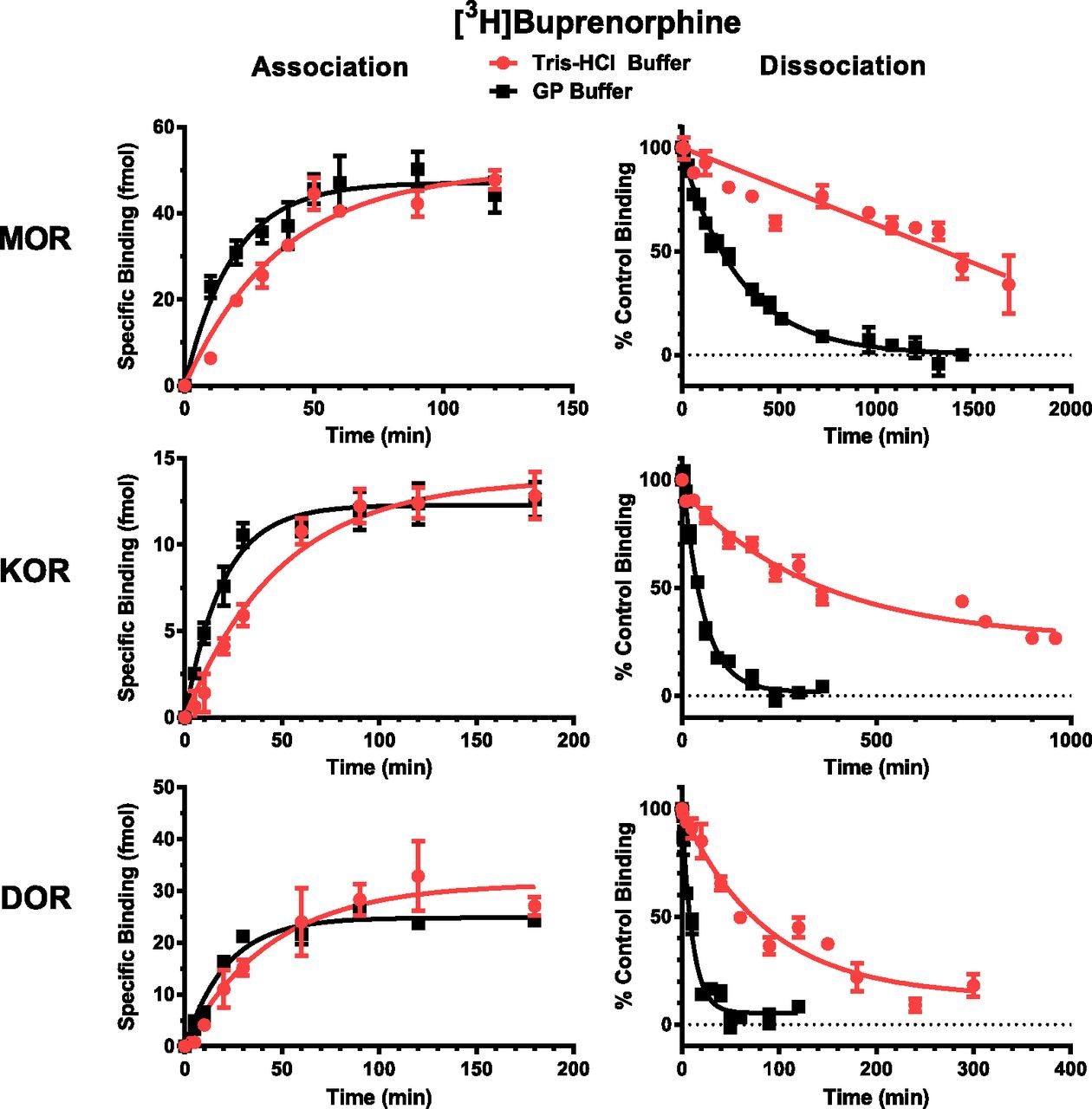
[3H]BUP bound specifically to the MOR with specific binding being 64% ± 3.5% and 73% ± 1.9% of total binding in Tris-HCl and GP buffers, respectively. Figure 2 shows the [3H]BUP association and dissociation time courses for the interaction of [3H]BUP with the MOR, KOR, and DOR performed in both the high-affinity Tris-HCl buffer and the low-affinity GP buffer. As reported in Table 3, the association T1/2 value for [3H]BUP with the multiple opioid receptors was approximately 2-fold faster in the GP buffer than in the Tris-HCl buffer. The dissociation rate for [3H]BUP was much faster in the GP buffer than in the Tris-HCl buffer. After reaching equilibrium, the dissociation of [3H]BUP from the receptor was initiated by the addition of 10 μM cold BUP. The dissociation T1/2 value for [3H]BUP was much longer at the MOR than at the KOR and DOR. In 50 mM Tris-HCl, pH 7.4, more than 23 hours was needed for 50% of the [3H]BUP to dissociate, whereas in the GP buffer 200 minutes resulted in half of the [3H]BUP being dissociated from the MOR. In Tris-HCl buffer, although more than 23 hours was needed for 50% dissociation from the MOR, 390 and 77 minutes were required for 50% dissociation from the KOR and DOR, respectively. In the GP buffer, the same rank order of rate of dissociation followed the results obtained with the Tris-HCl buffer, with the DOR having the fastest rate of dissociation, followed by the KOR, and then the MOR, which was five times slower than the KOR. Because of the slow dissociation of [3H]BUP from the MOR in the Tris-HCl buffer, it was not possible to measure the Koff, Kon, and Kd values. The rapid dissociation of [3H]BUP from the DOR in the GP buffer prohibited the accurate determination of the Kon and Kd values.

Association and dissociation of [3H]BUP with MOR, KOR, and DOR. Association assays were performed by incubating [3H]BUP with hMOR-CHO, hKOR-CHO, and hDOR-CHO cell membranes for various times in 50 mM Tris-HCl, pH 7.4, or GP buffer. Nonspecific binding was measured at each time point by inclusion of 10 μM naloxone. Dissociation assays were performed by incubating [3H]BUP with membranes for 90 minutes to reach equilibrium at 25°C and then adding 10 μM BUP at different times to displace the bound [3H]BUP. The [3H]BUP concentrations used are listed in Materials and Methods.
Association and dissociation T1/2 times for [3H]BUP and [3H]SAM binding at the MOR, KOR, and DOR in Tris-HCl and GP buffers
Data are the mean T1/2 values ± S.E.M. from three experiments performed in triplicate.
[3H]SAM had nonspecific binding of less than 1% when binding to the MOR in either Tris-HCl or GP buffers. Figure 3 shows that the association rate for [3H]SAM at the MOR was similar regardless of whether the Tris-HCl or GP buffer was used. In the GP buffer, the association rate for [3H]SAM at the KOR and DOR was fast, with the T1/2 values being less than 2 minutes (Table 3). Like [3H]BUP, [3H]SAM had the slowest dissociation from the MOR, with dissociation T1/2 values of 260 and 44 minutes in the Tris-HCl and GP buffers, respectively, after the addition of 1 μM cold SAM. Like the association rates at the KOR and DOR, the dissociation rates for [3H]SAM at these receptors were fast, with T1/2 values of less than 2 minutes in the GP buffers, whereas 47 and 20 minutes, respectively, were needed in the high-affinity Tris-HCl buffer. For equilibrium binding experiments, a 90-ml incubation was used with [3H]BUP and a 60-minute incubation was used with [3H]SAM.

Association and dissociation of [3H]SAM with MOR, KOR, and DOR. Association assays were performed by incubating [3H]SAM with hMOR-CHO, hKOR-CHO and hDOR-CHO cell membranes for various times in 50 mM Tris-HCl, pH 7.4 or GP buffer. Nonspecific binding was measured at each time point by inclusion of 10 μM naloxone. Dissociation assays were performed by incubating [3H]SAM with membranes for 60 minutes to reach equilibrium at 25°C and then adding unlabeled 1 μM SAM at different times to displace the bound [3H]SAM.
[3H]BUP and [3H]SAM Saturation Binding Experiments.
Figure 4 shows the results from saturation binding experiments for both [3H]BUP and [3H]SAM binding to the MOR in Tris-HCl and GP buffers. For both radioligands and binding conditions, a linear Scatchard plot was obtained. Similar results were obtained with the KOR and DOR as summarized in Table 4. [3H]BUP had the highest affinity for the KOR with Kd values of 0.14 ± 0.015 and 0.074 ± 0.011 nM in Tris-HCl and GP buffers, respectively. At the three opioid receptors, [3H]BUP had higher affinity when the binding assay was performed in GP buffer than in Tris-HCl buffer, which is like the results obtained in competition binding experiments (Table 2). [3H]SAM had the highest affinity for the MOR with Kd values of 0.046 ± 0.0027 and 0.044 ± 0.0051 nM in Tris-HCl and GP buffers, respectively. [3H]SAM had good affinity for both the KOR and DOR in addition to having a high affinity for the MOR (Table 4). Notably, Kd values did not show large shifts between the GP and Tris-HCl buffers, with the exception of a 16-fold decrease in [3H]SAM affinity for the KOR in the GP buffer. This decrease in the Kd value for [3H]SAM at the KOR agreed with the Kd value calculated from the association and dissociation data (Table 3).
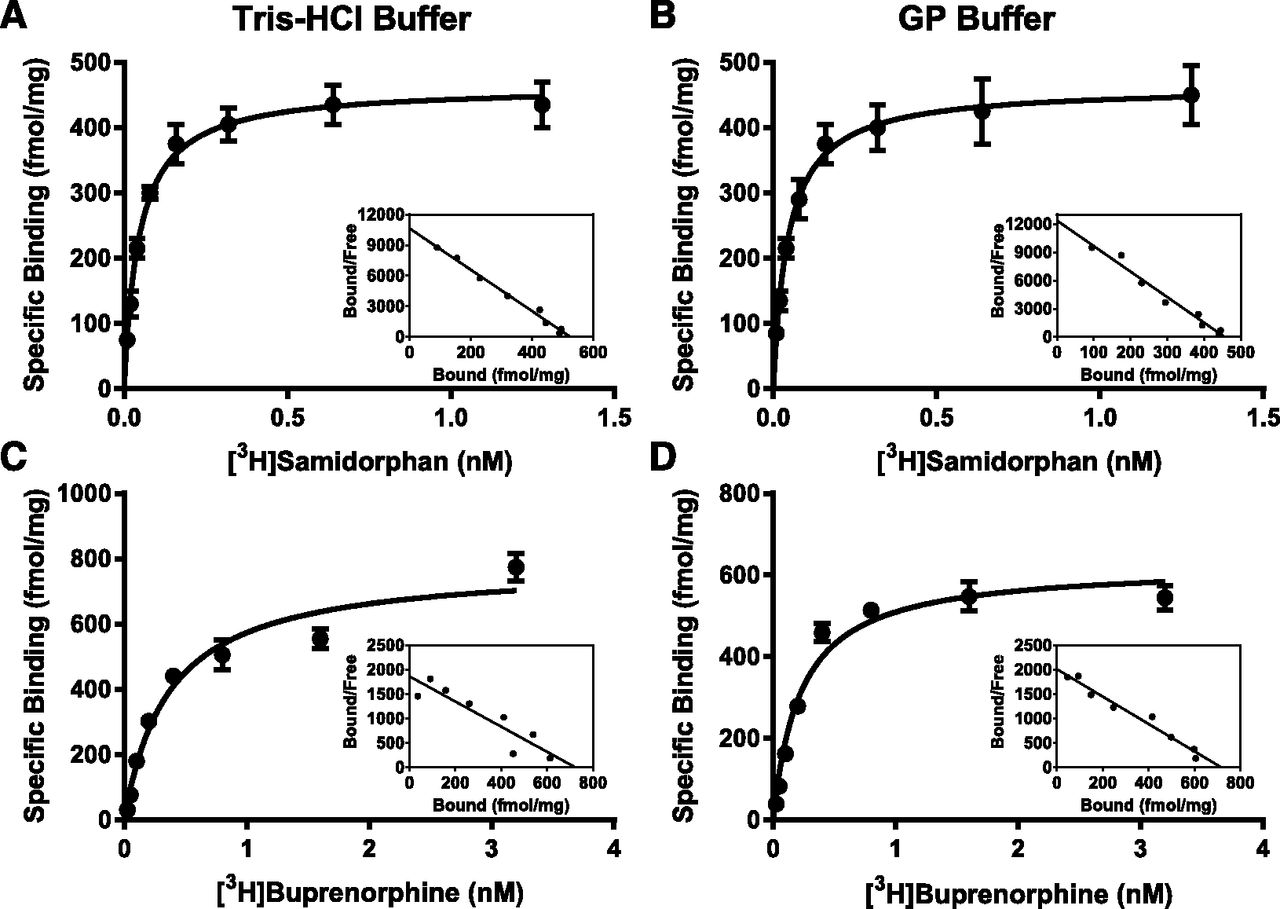
Saturation binding and representative Scatchard plots for [3H]SAM and [3H]BUP at the MOR in Tris-HCl or GP buffer. The binding affinity (Kd) and maximum binding (Bmax) of [3H]SAM (A and B) and [3H]BUP (C and D) under high- (A and C) and low-affinity binding conditions (B and D) to hMOR-CHO membranes. High-affinity binding was measured in 50 mM Tris-HCl buffer, pH 7.4. Low-affinity binding was measured in GP buffer. Eight concentrations of either [3H]BUP or [3H]SAM were incubated with hMOR-CHO membranes in 50 mM Tris-HCl, pH 7.4, or GP buffer at a final volume of 1 ml. Nonspecific binding was measured with 10 μM naloxone. hMOR-CHO membranes were added last. Data are the mean femtomoles bound per milligram of membrane protein ± S.E.M. A representative Scatchard plot is included for each condition.
Kd and Bmax values for [3H]BUP and [3H]SAM binding to the MOR, KOR, and DOR in Tris-HCl and GP buffers
Data are the mean Kd and Bmax values ± S.E.M. from at least three experiments performed in triplicate.
Bmax values for both [3H]BUP and [3H]SAM were a reflection of the number of receptors expressed in the CHO cell lines. It is not clear why more moles of [3H]BUP were bound to all three opioid receptors than was observed with [3H]SAM since the same membrane preparations were used with both 3H-labeled compounds.
Competition Binding Experiments with [3H]BUP and [3H]SAM at the MOR.
Table 5 shows that SAM, naloxone, and naltrexone competed with 0.4 nM [3H]BUP for binding to the MOR. SAM had the highest affinity with Ki values of 0.26 ± 0.014 and 0.24 ± 0.026 nM when the binding experiments were performed in Tris-HCl and GP buffers, respectively. Although SAM had a 5-fold higher affinity for the MOR when [3H]DAMGO was used as radioligand (Table 1), the Ki values for SAM in competing with [3H]BUP may be more representative of the pharmacology when the molecules are dosed as a combination in vivo. BUP, naloxone, and naltrexone were able to compete with 0.05 nM [3H]SAM for binding to the MOR. Naltrexone and BUP had Ki values of less than 0.4 nM regardless of which buffer was used in the binding experiments.
Ki values for the inhibition of [3H]BUP and [3H]SAM binding to the MOR by SAM, BUP, naltrexone, and naloxone in Tris-HCl and GP buffers
Data are the mean Ki values ± S.E.M. from three separate experiments performed in triplicate.
[35S]GTPγS Binding Mediated by the MOR and KOR in the Presence of BUP and SAM Alone and in Combination.
BUP stimulated [35S]GTPγS binding at the MOR with an EC50 value of 0.63 ± 0.40 nM and an Emax value of 57% ± 5.5%, indicating that BUP was a potent partial agonist at the MOR (Fig. 5A; Table 6). BUP partially inhibited DAMGO-stimulated [35S]GTPγS binding (Fig. 5C). SAM did not stimulate [35S]GTPγS binding for the MOR (Fig. 5A), but SAM completely inhibited DAMGO-stimulated [35S]GTPγS binding (Fig. 5C; Table 6). SAM did not act as an inverse agonist at the MOR because SAM alone did not decrease basal [35S]GTPγS binding (Fig. 5A). An initial assessment of the pharmacology of the combination of BUP and SAM was conducted at 1:1, 1:5, and 1:50 molar BUP/SAM ratios by assessing [35S]GTPγS binding using membranes from hMOR- and hKOR-CHO cells. As shown in Fig. 5A and Table 6, increasing the relative amount of SAM in the ratio caused the expected decrease in the Emax value for the combination on the MOR with virtually no measureable activity present at the 1:50 ratio.

Stimulation and inhibition of [35S]GTPγS binding to the MOR and KOR by BUP and SAM alone and in combination. (A) Stimulation of [35S]GTPγS binding mediated by the MOR by BUP and SAM alone or various molar ratios of the compounds together. (B) Stimulation of [35S]GTPγS binding at the KOR by BUP, SAM, or various molar ratios of the compounds together. (C) Inhibition of agonist-stimulated [35S]GTPγS binding at the MOR by BUP or SAM. (D) Inhibition of agonist-stimulated [35S]GTPγS binding at the KOR by BUP and SAM. When SAM and BUP were tested together, the total molarity was plotted.
Efficacy and potency of BUP and SAM alone and in combination in stimulating [35S]GTPγS binding mediated by the MOR, KOR, and DOR
Data are the mean values ± S.E.M. from three experiments performed in triplicate except where noted. The Emax value is the percentage of maximal stimulation and Imax is the percentage of maximal inhibition.
At the KOR, both BUP and SAM were partial agonists (Fig. 5, B and D; Table 6). SAM was more efficacious than BUP at the KOR, with an Emax value of 56% ± 0.59%, whereas BUP had an Emax value of 25% ± 1.3%. Interestingly, both the 1:1 and 1:5 BUP/SAM ratios showed Emax values similar to BUP alone (25% for BUP alone, 29% for both 1:1 and 1:5 BUP/SAM), whereas an increase in Emax values was not observed until the 1:50 BUP/SAM (Emax = 40%). These data suggest that SAM was less effective competing for binding with BUP on the KOR, despite having similar EC50 values. Notably, a decrease in affinity at the KOR for SAM was observed under the GP binding conditions (Table 4) and under buffer conditions like those of the [35S]GTPγS assay.
BRET Assays to Study BUP and SAM Mediating the Coupling of the MOR, KOR, and DOR to Specific G Proteins.
Previous studies demonstrated that opioid receptors couple to Gαi/o proteins, because opioids inhibit cyclic AMP production in a pertussis toxin (PTX)-sensitive manner (Law et al., 2000). Using BRET we conducted a series of cellular assays in which the signaling of BUP and SAM was studied for coupling of a specific opioid receptor to specific Gα proteins (Audet et al., 2008). Figure 6 shows the MOR, KOR, and DOR signaling through the Gαi1, Gαi2, and Gαi3 proteins. BUP was a partial agonist for MOR signaling through all the Gαi proteins, with greater efficacy in signaling through Gαi1 and Gαi3 than in signaling through Gαi2 (Fig. 6A; Table 7). SAM was an antagonist when the MOR was signaling through the Gαi proteins (Fig. 6B; Table 7). The combination of BUP and SAM was assayed at a 1:3 molar ratio, which approximates the steady-state concentrations of both molecules in humans for the BUP/SAM combination in development for MDD. The efficacy of BUP at the three Gαi proteins was reduced by the inclusion of SAM, consistent with earlier [35S]GTPγS activity assessments. Figure 6 shows that both BUP and SAM mediated the activation of GαoA, GαoB, and Gαz by the MOR. At both GαoA and GαoB, BUP had Emax values of 87% and 89%, respectively, suggesting that BUP was almost a full agonist when the MOR was signaling through Gαo proteins (Fig. 6A; Table 8). When signaling through the Gαz protein, BUP had an Emax value of 92%, demonstrating that BUP was very efficacious when the MOR was signaling through the Gαz protein (Fig. 6A; Table 8). SAM was a partial agonist at the MOR when signaling through GαoA, GαoB, and Gαz proteins (Fig. 6B; Table 8). Spider plots of the pEC50 and Emax values for BUP, SAM, and the 1:3 molar ratio of BUP/SAM in activating the Gαi, Gαo, and Gαz proteins allow for easy comparison of potency and efficacy of BUP and SAM signaling through the MOR, KOR, and DOR interacting with one Gα subunit (Supplemental Fig. 1). These findings show that both BUP and SAM were able to activate the MOR to signal through Gαo and Gαz proteins and that the concentrations of BUP needed to signal through Gαo and Gαz proteins were like the concentrations needed to activate Gαi signaling (Table 7). In contrast, although SAM was an antagonist when the MOR was signaling through Gαi proteins, it was a partial agonist when the MOR was signaling through Gαo and Gαz proteins (Figs. 6 and Table 8). In all cases assessed at the MOR, the Emax value observed with SAM was significantly lower than that observed with BUP, consistent with the intermediate Emax values observed in the 1:3 combination. Interestingly, a similar pattern of activity was noted for all of the MOR antagonists we assessed. Naltrexone was a partial agonist at the MOR when the MOR was activating the Gαz and to a lesser degree the GαoB protein (Supplemental Fig. 2; Table 8). Naloxone had a similar profile as naltrexone (Table 8).

BRET studies showing BUP and SAM alone and in combination activating MOR (A–C), KOR (D–F), and DOR (G–I) signaling through Gαi1, Gαi2, Gαi3, GαoA, GαoB, and Gαz. Transfection of HEK293 cells with receptor plasmids was performed with 3 μg/ml of PEI as the transfection reagent, and 1.8 × 106 cells in T-75 cell culture flask. Forty-eight hours after transfection, cells were washed once with phosphate buffered saline, removed with trypsin, centrifuged at 1000g, and the pellet was resuspended in Tyrode-HEPES buffer. Cells (10,000 cells per well) were dispensed into plates using the automated BioTek Multi-Flo reagent dispenser, and incubated at room temperature for 50 minutes with varying concentrations of BUP and SAM alone and in combination or control opioids. Coelenterazine was added subsequently at a final concentration of 2 μM. Cells were incubated for an additional 10 minutes at room temperature, and BRET readings were captured using the Envision plate reader (filters: 400/70 nm, 515/20 nm; PerkinElmer). BRET signals were determined by calculating the ratio of light emitted by GFP-acceptor (515 nm) over light emitted by luciferase-donor (400 nm). For each compound/biosensor pair, the compound-induced BRET signals were normalized to the BRET signal obtained from DAMGO, U50,488, and SNC-80 for the MOR, KOR, and DOR, respectively. Maximal and minimal BRET signal is defined by 20 μM DAMGO/20 μM U50,488/20 μM SNC-80, or cells in the absence of agonists, respectively.
EC50 and Emax values for BUP and SAM alone and in combination in stimulating the activation of Gαi1, Gαi2, and Gαi3 mediated by the MOR, KOR, and DOR
EC50 values are expressed in total molarity when BUP and SAM were combined. Data are the mean values ± S.E.M. from six determinations.
EC50 and Emax values for BUP and SAM alone and in combination in stimulating the activation of GαoA, GαoB, and Gαz mediated by the MOR, KOR, and DOR
EC50 values are expressed in total molarity when BUP and SAM were combined. Data are the mean values ± S.E.M. from six determinations.
Both BUP and SAM were partial agonists when the KOR activated the Gαi proteins (Fig. 6, D and E; Supplemental Fig. 1; Table 7). SAM had higher efficacy than BUP, with Emax values ranging from 55% to 60% for all three Gαi proteins. When BUP and SAM were combined at a 1:3 molar ratio, the efficacy and potency of the combination closely matched that of BUP alone, suggesting SAM did not compete effectively with BUP as was observed in the [35S]GTPγS activity assessment (Table 6). At the KOR, both BUP and SAM were partial agonists when the KOR signaled through the GαoA and GαoB proteins (Fig. 6, D and E; Table 8). As with Gαi signaling, SAM had a greater efficacy at stimulating signaling through GαoA and GαoB than did BUP. Both compounds had higher efficacy when signaling through GαoB than GαoA. As in the observations with Gαi signaling, the combination of BUP and SAM at a 1:3 molar ratio was virtually identical to BUP alone when signaling through Gαo proteins (Fig. 6F; Table 8). When the KOR was signaling through Gαz proteins, both BUP and SAM were full agonists (Fig. 6, D and E; Table 8).
At the DOR, BUP was a partial agonist, with Emax values ranging from 36% for Gαi1 to 18% for Gαi2 (Fig. 6G; Table 7). SAM had Emax values of slightly less than 20% for Gαi1 and Gαi3 (Fig. 6H; Table 7). The efficacy of SAM at Gαi2 was too low to be measured. Both BUP and SAM were less potent at the DOR than they were at the MOR and KOR. The 1:3 ratio of BUP/SAM showed Emax values intermediate to the two molecules alone, suggesting they effectively compete with each other, though it should be noted, given their similar potency and efficacy on the DOR, that it was difficult to provide a definitive assessment (Fig. 6I; Table 8). At the DOR, both BUP and SAM were partial agonists when the DOR was signaling through GαoA, GαoB, and Gαz proteins (Fig. 6, G and H; Table 8).
The activation of the inward-rectifying K+ channel (Kir) in response to the opioid receptor activating Gαi2 and GαoA was studied and the results are reported in Table 9. The β−γ subunits from the G protein interact with the K+ channel to activate the channel. BUP was a partial agonist in activating Kir when signaling was through either Gαi2 or GαoA and the MOR or DOR. These results are consistent with the results obtained for BUP activating the Gαi2 and GαoA proteins (Fig. 6; Tables 7 and 8). At the KOR, both BUP and SAM stimulated Gαi2-Kir activity with Emax values of 14% and 32%, respectively. SAM did not activate Kir channels mediated by MOR signaling through Gαi2 or GαoA proteins. Because BUP had Emax values of 30% or less, the 1:3 combination of BUP/SAM reduced the Emax values to levels that were too low to measure. At the KOR, SAM activated Gαi2-Kir and GαoA-Kir with Emax values of 32% and 21%, respectively (Table 9). BUP was less efficacious than SAM at the KOR in signaling through Gαi2-Kir and GαoA-Kir (Table 9). BUP was able to signal through the DOR and both Gαi2 and GαoA to activate the Kir channel (Table 9). At the DOR, SAM did not signal through either Gαi2 or GαoA to activate the Kir channel. The results obtained with the activation of the Kir channel by the β-γ subunits from either Gαi2 or GαoA paralleled the results obtained with the measurements of directly activating Gαi2 and GαoA.
EC50 and Emax values for BUP and SAM alone and in combination in stimulating the activation of Gαi2-Kir, GαoA-Kir, and β-arrestin recruitment mediated by the MOR, KOR, and DOR
EC50 values are expressed in total molarity when buprenorphine and samidorphan were combined. Data are the mean values ± S.E.M. from six determinations.
Upon activating the MOR, BUP caused a partial recruitment of β-arrestin when the G protein-coupled receptor kinase 2 (GRK2) was included in the β-arrestin assay. BUP had an Emax value of 33%, while SAM showed no activity in the same assay (Fig. 7A; Table 9). The addition of SAM blocked β-arrestin recruitment by BUP rendering the 1:3 BUP/SAM combination inactive (Fig. 7A; Table 9), suggesting that this approach may block the deleterious effects associated with β-arrestin signaling. There was very little β-arrestin recruitment by either BUP or SAM at the KOR or DOR (Fig. 7, B and C).

Recruitment of β-arrestin to the MOR (A), KOR (B), and DOR (C) by BUP and SAM alone and in combination. Transfection with receptor, GRK2, and biosensor plasmids was performed as described in Materials and Methods. Cells (10,000 cells per well) were dispensed into plates using the automated BioTek Multi-Flo reagent dispenser, and incubated at room temperature for 50 minutes with varying concentrations of BUP and SAM alone and in combination or with control opioids. Coelenterazine was added subsequently at a final concentration of 2 μM. Cells were incubated for an additional 10 minutes at room temperature, and BRET readings were captured using the Envision plate reader (filters: 400/70 nm, 515/20 nm; PerkinElmer). BRET signals were determined by calculating the ratio of light emitted by GFP-acceptor (515 nm) over light emitted by luciferase-donor (400 nm). For each compound/biosensor pair, the compound-induced BRET signals were normalized to the BRET signal obtained from DAMGO, U50,488, and SNC-80 for the MOR, KOR, and DOR, respectively. Maximal and minimal BRET signal is defined by 20 μM DAMGO/20 μM U50,488/20 μM SNC80, or cells in the absence of agonists, respectively. Morphine was included as a control.
Discussion
A combination of BUP and SAM at a 1:1 dose ratio is currently in development as an adjunctive treatment of MDD (Ehrich et al., 2015; Fava et al., 2016). The studies herein were undertaken to describe the opioid pharmacology of both components individually as well as in combination. Ki and Kd determinations for BUP and SAM were conducted in both low- and high-affinity conditions. In aggregate, the data demonstrate SAM and BUP had the highest affinity for the MOR and KOR, followed by the DOR and relatively weak affinity for the NOP receptor. The findings with BUP agree with previous studies (Huang et al., 2001; Cami-Kobeci et al., 2011; Khroyan et al., 2015). Full and partial MOR agonists showed the expected decrease in affinity under low-affinity binding conditions in accord with previous studies (Simon and Groth, 1975; Emmerson et al., 1996; Selley et al., 2000). However, BUP had higher affinity in the low-affinity buffer, a behavior to date only observed with inverse agonists (Emmerson et al., 2004). Indeed, there was little difference in binding affinity between the low- and high-affinity binding conditions with either molecule with the exception of a 16-fold decrease in the Kd value for [3H]SAM on the KOR under the low-affinity binding conditions. This finding was consistent with SAM acting as a partial agonist at the KOR in the [35S]GTPγS binding and BRET assays. On the basis of kinetic studies, the difference in affinity could be attributed largely to an increase in the off rate. It is plausible that the addition of GDP promotes inactive conformation(s) of the receptor (Selley et al., 2000; Chabre et al., 2009) suggesting BUP binds with equal affinity to both active and inactive conformations of the receptor.
Our studies also demonstrated that the Ki value obtained for SAM varied depending on the radioligand used for the assessment. Ki values for SAM were in general higher when competing with [3H]BUP versus other radioligands across all assays and conditions. Although such shifts won’t affect rank order potency, they are important to consider when the two agents are administered in combination.
The [35S]GTPγS binding assays conducted for BUP and SAM paralleled the data obtained with the binding assessment, particularly under low-affinity conditions and replicated previous studies conducted with BUP and SAM (Huang et al., 2001; Wentland et al., 2009). BUP showed similar potency as both an agonist and antagonist for both the MOR and KOR, again supporting the observation that its binding affinity is not dramatically changed when binding the active or inactive form(s) of both receptors. Combination studies conducted with decreasing ratios of BUP/SAM demonstrated a decrease in the Emax value on the MOR at all ratios tested versus BUP, whereas the Emax value for BUP was not affected except at the 1:50 BUP/SAM ratio on the KOR. These data suggested that SAM effectively competed for binding with BUP on the MOR but not on KOR. This relative decrease in affinity is consistent with the shift in affinity observed with SAM when assessed in low-affinity buffer conditions. Finally, signaling downstream of the MOR, KOR, and DOR was assessed in intact HEK293 cells using BRET. This technique allowed the activity of BUP and SAM and the 1:3 BUP/SAM ratio to be assessed on individual G proteins as well as β-arrestin. The data suggest remarkable promiscuity for agonist signaling downstream of the opioid receptors. In general for both BUP and SAM, Emax values were observed to shift across different signaling partners, whereas EC50 values remained relatively unchanged. For example, BUP had Emax values as low as 12% for Gαi2 and as high as 92% signaling through Gαz. An increase in Emax values for BUP, on the MOR signaling through the GαoA over the Gαi1, had been reported previously (Saidak et al., 2006).
To estimate the in vitro pharmacology of clinical administration of BUP/SAM, a molar ratio of 1:3 BUP/SAM was examined in the same panel. These concentrations approximate the ratio of the two molecules observed in clinical studies at steady-state exposure. SAM attenuated BUP’s Emax values when signaling through any of the tested G proteins. Across all assessments conducted on the MOR, the combination showed decreases in Emax values compared with BUP alone, suggesting SAM effectively competes with, and antagonizes the activity of, BUP regardless of whether SAM functioned as an antagonist or partial agonist for the assay in question.
Notably, a similar pattern of activity across the G proteins was observed for other MOR antagonists besides SAM. Naltrexone and naloxone were antagonists when the MOR was coupled to Gαi proteins but they were partial agonists when the MOR was signaling through the Gαz and the GαoB proteins. Both compounds were most efficacious when the MOR was signaling through the Gαz protein. A previous BRET study showed that naloxone activated GαoA, GαoB, and Gαz proteins, but not Gαi proteins, when signaling through the MOR (Masuho et al., 2015). Given that neither naltrexone, naloxone, nor SAM appear to be abused or have analgesic activity mediated by the MOR, the in vivo pharmacologic activity of the observed Gαo and Gαz will require further study.
In our studies, BUP was a partial agonist at the KOR when the receptor was signaling through Gαi and Gαo proteins and a full agonist when the KOR was signaling through the Gαz protein. Previous BUP studies using [35S]GTPγS binding at the KOR provided highly variable results that characterized BUP as a partial agonist at the KOR (Huang et al., 2001; Wentland et al., 2009) or an inverse agonist (Grinnell et al., 2016). SAM was a partial agonist at the KOR, when signaling through Gαi and Gαo, and a full agonist signaling through Gαz proteins. The 1:3 BUP/SAM combination showed activity that was virtually indistinguishable from BUP alone (see Fig. 7), suggesting that, at concentrations of BUP/SAM observed in blood clinically, SAM does not effectively compete for binding with BUP at the KOR regardless of the signaling pathway analyzed. These results are fully consistent with the [35S]GTPγS experiments in this study.
Previous studies found no recruitment of β-arrestin to the MOR by BUP (McPherson et al., 2010; Chen et al., 2013; Grinnell et al., 2016). In this study, BUP had an Emax value of 33% for β-arrestin recruitment to the MOR in HEK293 cells transfected with GRK2. Like results have been reported for PMZ21 and oliceridine 130 (TRV130) with GRK2-transfected cells (Manglik et al., 2016). Both SAM and the 1:3 molar combination of BUP/SAM showed no measureable activity in the β-arrestin recruitment assay. It is not clear whether this lack of β-arrestin recruitment is the result of the partial reduction in the efficacy of BUP or if the combination specifically blocks β-arrestin recruitment to the MOR. Notably, the only biased MOR ligands reported to date are partial agonists (Madariaga-Mazón et al., 2017).
In summary, our studies provide the most complete characterization of the in vitro pharmacology of BUP, SAM, and combinations thereof published to date. The data demonstrate the unique property of BUP to retain similar binding potency across a myriad of assessments, showing similar potency when acting as an agonist or antagonist. Distinctive properties of BUP in comparison with other μ partial agonists include its slow dissociation from the MOR and its slightly higher affinity for the MOR in the presence of NaCl and GDP. These distinguishing pharmacological properties of BUP may account for its ability to retain similar potency across many signaling assessments. SAM largely functioned as an antagonist at the MOR and partial agonist at the KOR. In all systems tested, SAM effectively decreased BUP’s efficacy on the MOR but was ineffective in modulating the KOR efficacy of BUP. Interestingly, the loss of potency at the KOR was not observed under standard high-affinity buffer systems but was predicted when studies were conducted in low-affinity buffer conditions. The in vivo implications of the differing levels of signaling observed with different G proteins in our studies are unclear and warrant further study.
Assessment of the 1:3 BUP/SAM indicate the combination retains a low level of efficacy at the MOR but functions like BUP at the KOR. In addition, the combination demonstrates a functional bias versus β-arrestin signaling, whereas BUP alone does not. Modulation of the opioid system through the MOR and KOR would be consistent with published studies demonstrating these targets may be of value in the treatment of MDD. For example, recent preclinical data suggest both MOR agonism and KOR antagonism could be of benefit depending on the behavioral domain assessed (Robinson et al., 2017). Although the current data does not address any contribution from potential metabolites of BUP or SAM though the relative potency of nor-BUP versus SAM and BUP at the MOR, a contribution of this metabolite seems improbable (Huang et al., 2001). Additional studies are currently underway to examine the effects of BUP/SAM in numerous behavioral models to verify the contributions of MOR and KOR in vivo and to better understand the interaction of opioid modulation with other neurotransmitter systems related to depression.
Acknowledgments
The authors thank Bruce Roth and Kelly Nichols for technical assistance and Ingo Mugge for his help in generating the spider plots.
Authorship Contributions
Participated in research design: Bidlack, Knapp, Deaver, Pin, Namchuk.
Conducted experiments: Knapp, Plotnikava, Arnelle, Wonsey, Fern Toh.
Performed data analysis: Bidlack, Knapp, Plotnikava, Arnelle, Wonsey, Fern Toh, Pin.
Wrote or contributed to the writing of the manuscript: Bidlack, Knapp, Pin, Fern Toh, Namchuk.
Footnotes
- Received April 9, 2018.
- Accepted August 1, 2018.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ALKS 5461
- the combination of buprenorphine and samidorphan at a 1:1 (mg/mg) dose ratio
- assay buffer
- 50 mM Tris-HCl, 3 mM MgCl2, 100 mM NaCl, 0.2 mM EGTA, pH 7.4
- BRET
- bioluminescence resonance energy transfer
- BUP
- buprenorphine
- CHO
- Chinese hamster ovary
- DAMGO
- [D-Ala2,NMePhe4-Gly-ol5]enkephalin
- DOR
- δ-opioid receptor
- GDP
- guanosine diphosphate
- GP buffer
- 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, and 50 μM GDP
- GRK2
- G protein-coupled receptor kinase 2
- HEK293
- human embryonic kidney 293
- Kir
- inward-rectifying K+ channel
- KOR
- κ-opioid receptor
- MDD
- major depressive disorder
- membrane buffer
- 50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4
- MOR
- μ-opioid receptor
- NOP
- nociceptin/orphanin FQ
- PEI
- polyethyleneimine
- PZM21
- 1-[(2S)-2-(dimethylamino)-3-(4-hydroxyphenyl)propyl]-3-[(2S)-1-(thiophen-3-yl)propan-2-yl]urea
- SAM
- samidorphan, (3-carboxyamido-4-hydroxy naltrexone)
- SNC-80
- (+)-4-[(α R)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3- methoxybenzyl]-N,N-diethylbenzamide
- U50,488
- 2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide
- U69,593
- (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}