


Visual Overview

Abstract
The inhibition of nicotinic acetylcholine receptors (nAChRs) has been proposed as a potential strategy to develop new antidepressant drugs. This is based on the observation that antidepressants that selectively block noradrenaline (NA) or serotonin (5-HT) reuptake also inhibit nAChRs. Dual antidepressants blocking both NA and 5-HT reuptake were proposed to shorten the delay in exerting their clinical effects; whether duloxetine, a prototype of dual antidepressants, also blocks nAChRs is unknown. Here we explored this question in bovine chromaffin cells (BCCs) that express native α3, α5, and α7 nAChRs and in cell lines expressing human α7, α3β4, or α4β2 nAChRs. We have found that duloxetine fully blocked the acetylcholine (ACh)-elicited nicotinic currents in BCCs with an IC50 of 0.86 µM. Such blockade seemed to be noncompetitive, voltage dependent, and partially use dependent. The ACh-elicited membrane depolarization, the elevation of cytosolic calcium ([Ca2+]c), and catecholamine release in BCCs were also blocked by duloxetine. This blockade developed slowly, and the recovery of secretion was also slow and gradual. Duloxetine did not affect Na+ or Ca2+ channel currents neither the high-K+–elicited [Ca2+]c transients and secretion. Of interest was that in cell lines expressing human α7, α3β4, and α4β2 nAChRs, duloxetine blocked nicotinic currents with IC50 values of 0.1, 0.56, and 0.85 µM, respectively. Thus, in blocking α7 receptors, which are abundantly expressed in the brain, duloxetine exhibited approximately 10-fold to 100- fold higher potency with respect to reported IC50 values for various antidepressant drugs. This may contribute to the antidepressant effect of duloxetine.
Introduction
Monoaminergic-based antidepressant therapies that activate either 5-HT or noradrenaline (NA) pathways have a latency of 2–3 weeks to respond and only approximately 70%–80% of patients are responders (Roose et al., 1986). To overcome these limitations, clinical studies suggest that combination therapy with drugs that inhibit both NA and 5-HT reuptake [serotonin and noradrenaline reuptake inhibitors (SNRIs)], may improve therapeutic efficacy and possibly could induce a faster onset of antidepressant activity compared with drugs that inhibit only one monoamine uptake system (Weilburg et al., 1989; Nelson et al., 1991; Seth et al., 1992). Inspired by this combination hypothesis, agents endowed with the ability to block both 5-HT and NA uptake such as duloxetine (Wong et al., 1993; Wong, 1998) have been developed.
The inhibition of nicotinic acetylcholine receptors (nAChRs) has been also proposed as a potential strategy to develop new antidepressant drugs. This is based in the cholinergic/adrenergic hypothesis, which postulated that hyperactivity of the cholinergic system contributes to depression (Janowsky et al., 1972). The findings supporting this hypothesis include the mood-improving effect of mecamylamine, a nonselective nAChR antagonist in patients with treatment-resistant depression, as well as the antidepressant-like activity of several nAChR antagonists and partial agonists in rodent models. In this line is the reported association between depression and smoking, likely due to desensitization of nAChRs by nicotine (Shytle et al., 2002; Mineur and Picciotto, 2010; Philip et al., 2010).
Also consistent with this cholinergic hypothesis is the observation that a number of key antidepressants such as the selective serotonin reuptake inhibitors fluoxetine, sertraline, paroxetine, and citalopram; the selective NA reuptake inhibitor reboxetine; the NA-dopamine reuptake inhibitor bupropion; and tricyclics amitriptyline, imipramine, and nortriptyline, have all been shown to possess antagonistic activities at nAChRs (Hennings et al., 1997; Fryer and Lukas, 1999a; López-Valdés and García-Colunga, 2001; Shytle et al., 2002; Arias et al., 2006; Bianchi, 2008; Arias et al., 2010a,b). On the other hand, nicotine itself and some nicotinic agonists or antagonists, can potentiate the antidepressant-like effects of selective serotonin reuptake inhibitors and selective noradrenaline reuptake inhibitors in rodent models of depression (Popik et al., 2003; Andreasen et al., 2011).
Duloxetine is a dual antidepressant that binds selectively with high affinity to both NA and 5-HT transporters (Karpa et al., 2002) and equipotently inhibits the reuptake of [3H] 5-HT and [3H] NA (Kasamo et al., 1996). From a clinical point of view, it has been found that duloxetine behaves as an effective acute and long-term treatment for patients with major depressive disorder (Detke et al., 2002; Goldstein et al., 2002; Perahia et al., 2006). Additionally, pooled analyses and meta-analyses have also reported the comparative efficacy and safety of duloxetine compared with other antidepressants in patients with major depressive disease (Gartlehner et al., 2009).
To our knowledge, whether duloxetine blocks nAChRs is unknown. Inasmuch as such a blockade could contribute to its efficacy as exemplified by the various antidepressants discussed above and may lead to adverse effects, we planned this study to find out whether duloxetine affected the nAChRs currents, other voltage-dependent ion currents, membrane excitability, Ca2+ signaling, and exocytosis in adrenal bovine chromaffin cells (BCCs). These cells are known to express the ganglionic type nAChRs of the α3β4 and α5 subtypes (Campos-Caro et al., 1997) as well as α7 nAChRs (López et al., 1998; Fuentealba et al., 2004). When stimulated by endogenously released acetylcholine (ACh), the physiologic neurotransmitter at the splanchnic nerve-chromaffin cell synapse, the stimulation of nAChRs by ACh causes a sudden discharge of catecholamine that is entirely dependent on Ca2+ entry through voltage-activated calcium channels (VACCs) (Douglas and Rubin, 1961; García et al., 2006), which are recruited as a result of ACh-elicited membrane depolarization (de Diego et al., 2008). We have found here that duloxetine causes a time- and concentration-dependent blockade of the whole-cell inward ACh-elicited current [inward current through nicotinic receptors (IACh)] in voltage-clamped BCCs. This blockade was exerted with different potencies on the subtypes of human α7, α3β4, and α4β2 nAChRs expressed in cell lines. The blockade of IACh in BCCs is accompanied by the inhibition of ACh-elicited cytosolic calcium ([Ca2+]c) transients and secretion responses; Na+ and Ca2+ channel currents, as well as the secretory responses triggered by high K+ were unaffected, indicating that duloxetine did not target the VACCs of BCCs.
Materials and Methods
Isolation and Culture of Bovine Chromaffin Cells.
Adrenal glands were obtained from a local slaughterhouse under the supervision of the local veterinary service. Chromaffin cells were isolated by digestion of the adrenal medulla with collagenase following standard methods with some modifications (Moro et al., 1990). Cells were suspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 50 IU/ml penicillin and 50 μg/ml streptomycin. To mitigate fibroblast growth, cells were plated in a medium containing 10 μM cytosine arabinoside, 10 μM fluorodeoxyuridine, and 10 μM leucine methyl ester. For catecholamine release measurements in chromaffin cell populations, cells were plated on 5-cm-diameter Petri dishes at 5 × 106 cells/dish. For patch-clamp studies, cells were plated on 1-cm-diameter glass coverslips (Labbox, Barcelona, Spain) at low density (5 × 104 cells/coverslip). For the study of changes in [Ca2+]c, cells were plated on 2.5-cm-diameter glass coverslips (Menzel Gläser; VWR, Barcelona, Spain) at a density of 15 × 104 cells/coverslip. Cultures were maintained in an incubator at 37°C in a water-saturated atmosphere with 5% CO2. Cells were used 1–4 days after plating. All experiments in this study were performed at room temperature (24 ± 2°C).
Culture of Cell Lines Overexpressing α7, α3β4, or α4β2 nAChRs.
The cell lines overexpressing human α7 (SH-SY5Y-hRα7), α3β4 (HEK293-hRα3β4), and α4β2 (HEK293-hRα4β2) and cell culture procedures were provided by Novartis Pharma AG (Basel, Switzerland). Briefly, HEK293-hRα3β4 and HEK293-hRα4β2 cells were cultured in a 1:1 mixture of DMEM containing 3.7 g/l NaHCO3 and 1.0 g/l glucose, supplemented with l-glutamine (524 mg/l), and Ham’s F-12 Nutrient Mixture containing 1.176 g/l NaHCO3 and supplemented with 10% (v/v) FBS, geneticin (0.2 mg/ml), and hygromycin B (0.2 mg/ml). SH-SY5Y-hRα7 cells were cultured in DMEM supplemented with 10% (v/v) FBS and 100 μg/ml geneticin. All cells were cultured at 37°C in 5% CO2 at 95% relative humidity and passaged every 3 days by being detached from the cell culture flask by being washed with DMEM and a brief incubation (∼3 minutes) with trypsin (0.5 mg/ml) and EDTA (0.2 mg/ml).
For studies of patch clamp at the single-cell level, cells were plated on 1-cm-diameter glass coverslips (Labbox) at low density (4 × 104 cells/coverslip) and used 1–4 days after plating. These experiments were carried out at room temperature (24 ± 2°C).
Whole-Cell Current Recordings.
Inward currents through nicotinic receptors (IACh), through voltage-activated sodium channels (INa), and through VACCs (ICa) were recorded using the voltage-clamp mode of the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). Whole-cell recordings were made with fire-polished borosilicate pipettes (resistance, 4–8 MΩ) that were mounted on the headstage of an EPC-9 patch-clamp amplifier (HEKA Electronik, Lambrecht, Germany), allowing cancellation of capacitative transients and compensation of series resistance. Data were acquired with a sample frequency of 20 kHz by using PULSE version 8.74 software (HEKA Elektronik). Data analyses were performed with PULSE version 8.74 programs (HEKA Elektronik). Coverslips containing the cells were placed on an experimental chamber mounted on the stage of a Nikon (Tokyo, Japan) Diaphot inverted microscope. Cells were continuously superfused with a control Krebs-HEPES solution composed of the following (in millimolar): 145 NaCl, 1.2 MgCl2, 5.6 KCl, 2 CaCl2, 10 HEPES, and 11 glucose (pH 7.4, NaOH).
To monitor IACh, INa, and ICa, cells were internally dialyzed with an intracellular solution containing as follows (in millimolar): 100 Cs-glutamate, 14 EGTA, 20 TEA-Cl, 10 NaCl, 5 Mg-ATP, 0.3 Na-GTP, and 20 HEPES (pH 7.3, CsOH). The external solutions were rapidly exchanged using electronically driven miniature solenoid valves coupled to a homemade multibarrel concentration clamp device (Gonzalez-Rubio et al., 2006; Lopez-Gil et al., 2017), the common outlet of which was placed within 100 μm of the cell to be patched. The flow rate was 1 ml/min and was regulated by gravity, allowing the complete replacement of solution surrounding the cell under experimentation in less than 50 milliseconds.
To measure all different currents, cells were held at −80 mV. In BCCs as well as in cell lines expressing human α3β4 or α4β2 nAChRs, IACh was generated by the application of 250-millisecond pulses of ACh (100 μM) every 30 seconds; in the case of the cell line expressing human α7 nAChRs, PNU120596 at 10 µM was used as an allosteric modulator of the receptor and PNU282987 at 10 µM as an agonist. The modulator was continuously perfused and PNU282987 was applied for 500 milliseconds every 60 seconds. In BCCs, ICa was generated by 50-millisecond depolarizing pulses from −60 to +60 mV every 20 seconds with 10 mV steps; INa was obtained with the same protocol as the one used for ICa. To monitor the resting membrane potential (Vm) in BCCs, recordings were made under the current-clamp mode of the patch-clamp technique. The pipette-filling solution contained the following (in millimolar): 120 K-glutamic acid, 14 EGTA, 10 NaCl, 5 Mg-ATP, 0.3 Na-GTP, and 20 HEPES (pH 7.3, KOH). All experiments were performed at room temperature (24 ± 2°C) on cells from 1 to 4 days after culture.
Measurement of [Ca2+]c in Single BCCs with Fura-2 AM.
To monitor the changes of [Ca2+]c, BCCs were incubated in DMEM containing the Ca2+ probe Fura-2/acetoxymethyl ester (Fura-2/AM) (10 μM, 1 hour at 37°C). After this incubation period, the glass coverslips containing the cells were mounted in a chamber, washed, and covered with Tyrode solution composed of the following (in millimolar): 137 NaCl, 1 MgCl2, 5.3 KCl, 2 CaCl2, 10 HEPES, and 10 glucose (pH 7.4, NaOH). The high K+ solutions were prepared by replacing equiosmolar concentrations of NaCl with KCl. The setup for fluorescence recordings was composed of a Leica DMI 4000 B inverted light microscope (Leica Microsystems, Barcelona, Spain) equipped with an oil-immersion objective (Leica 40× Plan Apo; numerical aperture, 1.25). The cells were continuously superfused by means of a five-way superfusion system at 1 ml/min with a common outlet 0.28-mm tube driven by electrically controlled valves with Tyrode solution. Fura-2/AM was excited alternatively at 340 ± 10 and 387 ± 10 nm using a Küber CODIX Xenon 8 Lamp (Leica Microsystems). Emitted fluorescence was collected through a 540 ± 20 nm emission filter and measured with an intensified charge-coupled device camera (camera controller C10600 ORCA R2; Hamamatsu Photonics K.K., Hamamatsu City, Japan). Fluorescence images were generated at 1-second intervals. Images were digitally stored and analyzed using LAS AF Software (Leica Microsystems). At the time of experiment performance, proper amounts of drug stock solutions were freshly dissolved into the Tyrode solution. All experiments were performed at room temperature (24 ± 2°C) on cells from 1 to 4 days after culture.
Online Amperometric Recording of Burst Catecholamine Release from Populations of BCCs.
Cells were scrapped off carefully from the bottom of the Petri dish (5 × 106 cells per dish) with a rubber policeman and centrifuged at 120g for 10 minutes. The cell pellet was resuspended in 200 μl of Krebs-HEPES solution. Cells suspended were trapped in a microchamber and perifused at a rate of 2 ml/min. The liquid flowing from the perifusion chamber reached an electrochemical detector model VA 641 (Metrohm, Herisau, Switzerland) placed just at the outlet of the microchamber, which monitors online the amount of catecholamine secreted under the amperometric mode. This amperometric strategy permits the online recording of reproducible catecholamine release responses during long time periods of 30–60 minutes. Catecholamines are oxidized at +650 mV, and the oxidation current was recorded on a PC computer (Borges et al., 1986).
Data and Statistical Analysis.
Data analysis of amperometric recordings in chromaffin cell populations were performed using Origin Pro software (version 8.6; OriginLab Corporation, Northampton, MA). Data analysis of ion currents and cell excitability was performed with PULSE version 8.74 programs (HEKA Elektronik). Fluorescent images were analyzed using LAS AF Software (Leica Microsystems). All statistical analysis and graph representations were performed using GraphPad Prism software (version 5.01; GraphPad Software, San Diego, CA). Data are expressed as the mean ± S.E.M. Statistical significance was established at P values smaller than 0.05. Post hoc tests were performed only if P < 0.05, and there was no significant variance in homogeneity.
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials.
Acetylcholine (catalog no. A6625), duloxetine (catalog no. Y0001453), and mecamylamine (catalog no. M9020) were purchased from Sigma-Aldrich (Madrid, Spain); PNU120596 (catalog no. 2498/5) and PNU282987 (catalog no. 2303/10) were purchased from Tocris Bioscience (Madrid, Spain).
Results
Effects of Duloxetine on IACh in BBCs.
The central question of this study was whether duloxetine blocked nAChRs. We tested this in voltage-clamped BCCs that were intermittently challenged with brief ACh pulses to elicit inward whole-cell nicotinic currents that are known to be mostly carried by sodium (Fenwick et al., 1982).
Figure 1A shows a prototype experiment performed in an example BBC whereby the first ACh pulse generated a current trace of 1.7 nA. In subsequent ACh pulses, IACh amplitudes underwent no decay. Thus, this protocol resulted in the ability to explore, within the same cell, the effect of a single or various concentrations of duloxetine, as exemplified in the original superimposed IACh traces displayed in Fig. 1B. Note the gradual current blockade elicited by 1–10 µM duloxetine.
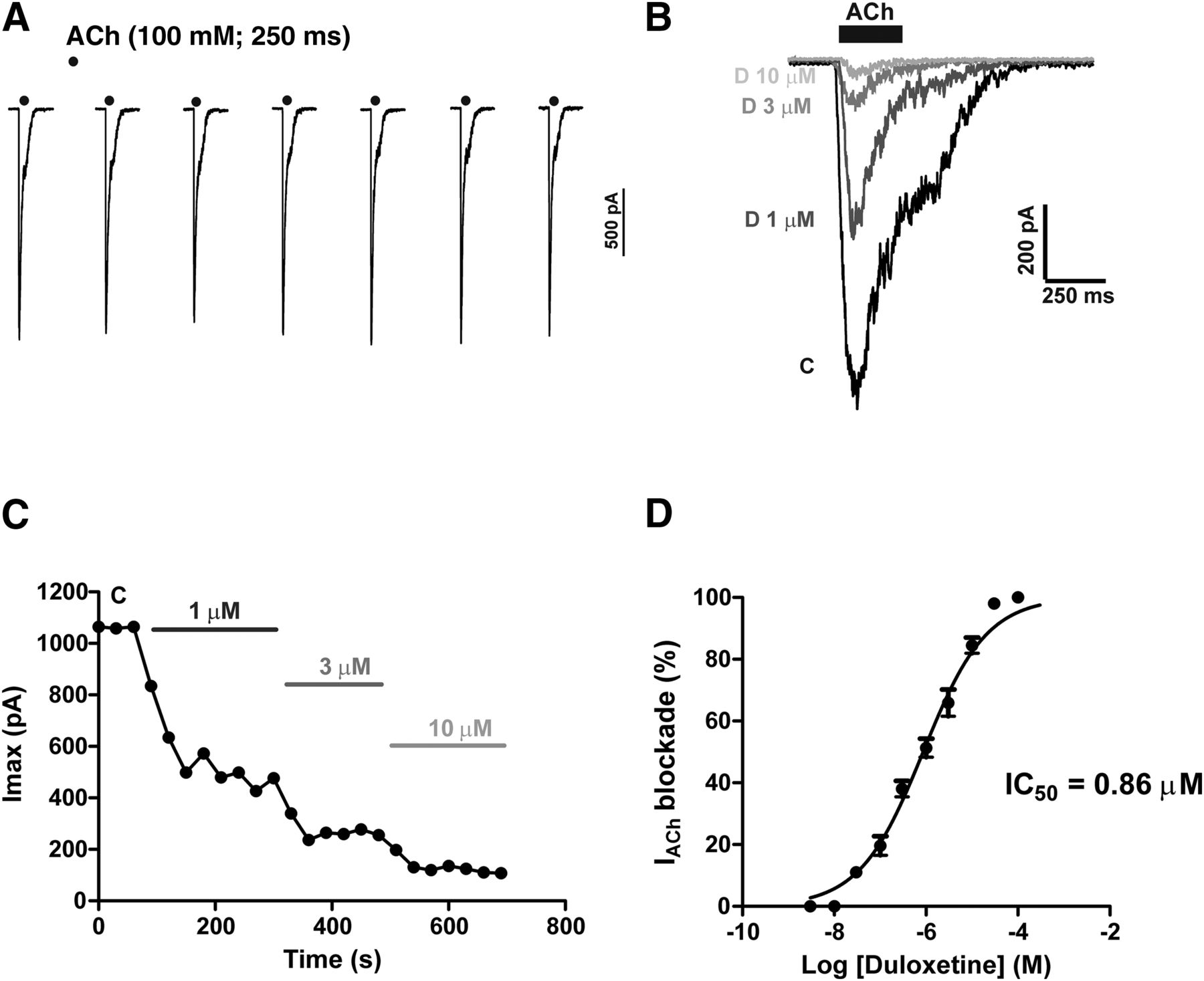
ACh-evoked whole-cell inward currents (IACh) are blocked by duloxetine in a concentration-dependent manner. BCCs were voltage clamped at −80 mV under the whole-cell configuration of the patch-clamp technique. The cell was continuously perifused with Krebs-HEPES solution. IACh currents were generated with 250-millisecond pulses of ACh (100 µM) applied at 30 second intervals. (A) Sequence of IACh traces obtained from an example cell, showing no decay of peak current amplitude. Example IACh traces obtained from a different cell (B) before adding duloxetine [control current (C)] and in the presence of increasing concentrations of duloxetine (D) (each trace was obtained after 4–5 minutes of cell perifusion with the drug). (C) Time course of IACh inhibition by increasing concentrations of duloxetine, added during the time periods indicated by the horizontal bars. Note the gradual blockade exerted by all duloxetine concentrations. (D) Concentration-response curve of IACh blockade by duloxetine, with an estimated IC50 value of 0.86 µM. Data are the mean ± S.E.M. of 10–17 cells.
A notable property of IACh blockade elicited by duloxetine was its gradual development that stabilized in a novel lower current plateau only after three ACh pulses (i.e., in about 1.5–2 minutes) (Fig. 1C). This may concord with the blockade by duloxetine of the ACh-elicited catecholamine release, which, as described later, was also gradual. A concentration-response curve of IACh blockade gave a calculated IC50 value of 0.86 µM (Fig. 1D).
IACh Blockade Exerted by Duloxetine in Resting BCCs.
Whether duloxetine binds to its site on the nAChR ion pore in its open or closed conformation, was tested with the following protocol. Cells voltage clamped at −80 mV were subjected to two periods of ACh stimulation, one initial period to determine the size of IACh peak amplitude (control current) and a second period after cells were exposed to duloxetine under resting conditions for 2 minutes (duloxetine perfusion started 30 seconds after the last ACh control pulse) (Fig. 2A). With this protocol, it was possible to discern whether at a given concentration, duloxetine was capable of producing a blockade of IACh that was similar to that elicited under repeated cell stimulation with ACh.

Blockade by duloxetine of ACh-elicited currents (IACh) is partially use dependent. IACh was elicited by the application of 250-millisecond pulses of 100 µM ACh to BCCs voltage clamped at −80 mV. (A) The protocol used. Cells were initially challenged with an ACh pulse to generate the initial control current (C). Then, the resting cell was exposed to 3 µM duloxetine during a 2-minute period. Thereafter, stimulation with ACh pulses applied at 30-second intervals was initiated, always in the presence of duloxetine. (B) Superimposed IACh traces obtained in a cell first challenged with ACh; then, duloxetine at 3 µM was added for 2 minutes and ACh stimulation was again restarted. (C) Pooled data on the blockade achieved by ACh pulses P1 to P7. Data are the mean ± S.E.M. of the number of cells shown in parentheses. Cells are from three different cultures.
The superimposed IACh traces of Fig. 2B were taken from a cell subjected to the protocol shown in Fig. 2A. The ACh pulse 1 (P1), applied after 2 minutes of cell exposure to 3 µM duloxetine, generated an IACh that was blocked by 40% with respect to the control initial current. During the subsequent ACh pulses (P2 to P7), applied in the presence of duloxetine, an additional gradual blockade was produced. Quantitative data on the blockade exerted by duloxetine are displayed in Fig. 2C. The higher fraction of blockade was achieved at P1, although some additional nonsignificant blockade was observed at pulses P2 to P7.
Blockade by Duloxetine of IACh at Different Holding Potentials and at Increasing ACh Concentrations.
These experiments were designed to test whether duloxetine caused a blockade of IACh in a voltage-dependent manner. BCCs were voltage clamped at holding potentials (HPs) of −100, −80, −60, or −40 mV and experiments similar to those displayed in Fig. 1 were performed. Duloxetine was used at 3 µM.
Figure 3A shows paired traces obtained from different cells at the HPs indicated on top. Note the drastic IACh blockade elicited by duloxetine at the four HPs tested. Pooled data on current blockade displayed in Fig. 3B indicate a similar degree of IACh blockade at HPs of −100 and −80 mV (71.73% ± 3.343% and 65.85% ± 4.50%, respectively). However, at −60 and −40 mV a significant lesser blockade occurred (51% ± 3.03% at −60 mV; 45.40% ± 4.15% at −40 mV), indicating that duloxetine blockade of IACh exhibits some voltage dependency.

Blockade by duloxetine of IACh at different HPs and at increasing ACh concentrations. BCCs were voltage clamped at different HPs (different cells were used for each HP) under the whole-cell configuration of the patch-clamp technique. IACh currents were generated with 250-millisecond pulses of ACh (100 µM) applied at 30-second intervals. After current stabilization, duloxetine (Dulox) 3 µM was perfused until a steady-state blockade was accomplished. (A) Superimposed IACh traces from example cells that were voltage clamped at the HPs shown on top of each pair of traces, recorded in the absence of [control (C)] and after 3-minute exposure to duloxetine 3 µM. (B) Pooled data on the blockade of IACh (normalized as the percentage control, ordinate) at the different HPs tested (bottom at each column). (C) Blockade of IACh elicited by 3 μM Dulox using increasing ACh concentrations (abscissa), in cells voltage clamped at −80 mV; currents were generated by 250-millisecond pulses of ACh applied at 30-second intervals. Data in (B) and (C) are represented as the mean ± S.E.M. of the number of cells from three different cultures shown in parentheses. *P < 0.05; **P < 0.005 with respect to the IACh blockade exerted by duloxetine at −80 mV HP (one-way analysis of variance, Dunnett’s multiple-comparison test).
Experiments were also performed to test the degree of IACh blockade by duloxetine, at increasing ACh concentrations. Figure 3C shows that in 15 cells, duloxetine causes 63% blockade of IACh; at 300 and 1000 μM, the extent of IACh blockade was also around 60%.
Duloxetine Blocks the Depolarizing Effect of ACh.
The physiologic secretory effects of ACh in the adrenal medulla are mediated by depolarization of chromaffin cells and recruitment of VACCs (de Diego et al., 2008). It was therefore expected that the blockade by duloxetine of IACh under the voltage-clamp mode of the patch-clamp technique (Figs. 1–3) could also block the ACh-elicited cell depolarization, which we explored under the current-clamp mode.
Figure 4A shows a record of the Vm of a current-clamped BCC. The initial Vm at approximately −75 mV was quite stable during the initial 2-minutes recording period. Duloxetine at 10 µM did not alter the resting Vm during a 3-minute exposure. ACh (30 seconds at 100 µM) added on top of duloxetine caused a transient depolarization followed by fast Vm recovery toward basal levels, indicating a full blockade of the ACh depolarization in the presence of 10 µM duloxetine. The blockade of ACh depolarization by duloxetine was better revealed in the experiment of Fig. 4B, consisting of an initial 30-second challenge with ACh that produced a Vm shift from −75 mV to a more or less stable new level of Vm at around −22 mV. Duloxetine was then applied, and after 70 seconds a new ACh pulse produced a transient depolarization that returned to resting Vm levels, indicating once more a practically full blockade of the depolarizing effects of ACh. In the experiment shown in Fig. 4C, the cell was exposed to ACh for 15 seconds, which generated a 30-mV depolarization with a sustained burst of action potentials. Added on top of ACh, duloxetine (for another 15 seconds) immediately suppressed the action potential firing and restored Vm to basal values in about 5–10 seconds. In the experiment shown in Fig. 4D, the classic blocker of nAChR mecamylamine at 10 µM, also blocked the depolarizing actions of ACh. Pooled data are presented in Fig. 4E; ACh reduced Vm from −79.04 ± 3.64 (19 cells) to −28.27 ± 2.62 mV (18 cells); such a depolarizing effect was fully blocked by 10 µM duloxetine (11 cells) as well as by 10 µM mecamylamine (eight cells).

Duloxetine blocks the depolarizing effects of ACh. The resting Vm and the depolarizing effects of ACh were explored under the whole-cell configuration of the patch-clamp technique, using the current-clamp mode. (A) Example trace showing the lack of effect of duloxetine (10 µM) on resting Vm, and blockade of ACh (100 µM) elicited depolarization by duloxetine. (B) Example Vm trace showing the depolarizing effect of ACh and its blockade by duloxetine. (C) Sustained depolarizing effect of ACh with superimposed action potentials and their fast suppression by adding duloxetine on top of ACh (bottom horizontal bars). (D) Example Vm trace showing the depolarizing effect of ACh and its blockade by 10 µM mecamylamine. (E) Pooled data on the depolarizing effects of ACh (white column) and the prevention of such depolarization by 10 µM each of duloxetine (Dulox) or mecamylamine (Mecam); data are the mean ± S.E.M. of the number of cells shown in parentheses from at least two different cell cultures. ***P < 0.001 with respect to Basal (one-way analysis of variance, Newman-Keuls multiple-comparison test).
Effects of Duloxetine on [Ca2+]c Transients Elicited by ACh.
ACh causes cell depolarization and action potentials (Fig. 4C), thereby augmenting Ca2+ entry through VACCs and the elevation of [Ca2+]c. Upon repeated ACh pulses (100 μM, 2 seconds) applied to Fura-2/AM–loaded BCCs, given at 2-minutes intervals to allow baseline recovery, the [Ca2+]c transient amplitudes were fairly constant along the nine pulses (not shown). Figure 5A displays a record averaged from 28 cells showing three [Ca2+]c transients of similar amplitudes evoked by P1 to P3 ACh pulses. Duloxetine (3 µM) applied beginning 2 minutes before P4 caused a pronounced blockade of the [Ca2+]c transients. At 2 minutes after duloxetine washout, the transient amplitude clearly recovered, although to a lower level than the initial P1–P3 responses. A more pronounced blockade was achieved with 10 µM duloxetine (∼75% blockade at P4), which further increased at P5 and P6 (Fig. 5B). The recovery after 2–6 minutes of drug washout was again rapid but partial. Normalized averaged data on [Ca2+]c transient amplitudes at P2 (control) and P5 (after a 4-minute exposure to duloxetine) are graphed in Fig. 5C; although the blockade reached 57.08% ± 2.04% at 3 µM (28 cells), at 10 µM duloxetine the blockade was augmented to 79.52% ± 1.24% (20 cells). These figures are in consonance with those of IACh and the secretion blockade exerted by duloxetine.

Duloxetine blocks the ACh-elicited [Ca2+]c transients in Fura-2/AM–loaded BCCs. Individual cells were challenged with nine sequential pulses of ACh (100 µM, 2 seconds; dots at the bottom of each transient), applied at 2-minute intervals to allow each transient to recover baseline level before applying the next pulse. Duloxetine (3 or 10 µM) was cell perifused 2 minutes before pulse P4 and during pulses P4 to P6. (A) [Ca2+]c transient traces (monitored as Fura-2/AM fluorescence ratios; calibration bar on the left) averaged from 28 cells from three different cultures, showing the blocking effects of 3 µM duloxetine (bottom horizontal bar). (B) Similar [Ca2+]c transient experiments, using 10 µM duloxetine. (C) [Ca2+]c transient peak amplitudes normalized as the percentage of control (ACh pulse P2 before duloxetine) and 4 minutes after duloxetine application (ACh pulse P5). Data are the mean ± S.E.M. of the number of cells and cultures shown in parentheses. ***P < 0.001 with respect to control P2 (unpaired t test).
Effects of Duloxetine on Catecholamine Release Elicited by ACh Pulses from Populations of BCCs.
The blockade of IACh and mitigation of ACh-evoked [Ca2+]c elevations predicts that duloxetine should also block the ACh-elicited catecholamine responses from BCCs. This was tested in BCC populations.
In BCC populations (5 million cells trapped in a perifusion microchamber), ACh (100 µM for 3 seconds) was applied at 1-minute intervals to elicit the release of catecholamines, which was monitored online with an amperometric detector. The initial three to four ACh pulses produced increasing secretory responses that were stabilized in the following 10–12 pulses. At this moment, the responses commenced a slow decay to stabilize at 30 minutes of stimulation at about 50%–60% of the initial responses, as shown in the example control trace of Fig. 6A. Duloxetine at 3 µM caused a gradual decrease of the secretory responses that stabilized only after 10 minutes or more of cell perifusion with the drug; upon drug washout, the responses also recovered very slowly (Fig. 6B, example experiment). Pooled data from several experiments, with the protocols shown in Fig. 6, A and B, are displayed in Fig. 6C. Note that the blockade of secretion elicited by increasing duloxetine concentrations lasted 10–15 minutes to reach steady state, and that this was true for all concentrations tested; also note the gradual recovery after washout that only reached the control responses after 10–15 minutes. Finally, a concentration-response plot of the steady-state blockade exerted by duloxetine at each concentration shows an IC50 value of 0.77 µM (Fig. 6D).

Duloxetine blocks the release of catecholamine from BBC populations challenged with ACh. Five million cells trapped in a microchamber and perifused with saline at 37°C were intermittently stimulated with 3-second ACh (100 µM) pulses given at 1-minute intervals [dots at the bottom of (A) and (B)]. Secretion responses were monitored online with an amperometric detector. (A) Example control trace from a batch of cells. (B) An example trace similar to that of (A), but here duloxetine at 3 µM was present, as indicated by the bottom horizontal bar. (C) Pooled results from experiments like those of (A) and (B). (D) Concentration-response curve for the blocking effects of duloxetine of ACh-elicited secretory responses; points were taken once steady-state secretion was reached, in the presence of duloxetine [arrow in (C)]. The IC50 value was calculated, and a value of 0.77 µM was obtained. Data are the mean ± S.E.M. of the number of cell batches and different cell cultures shown in parentheses. ***P < 0.001 with respect to P5 of control pulses (one-way analysis of variance, Dunnett’s multiple-comparison test).
Effects of Duloxetine on Na+ and Ca2+ Currents.
The sequence of events occurring at the splanchnic-chromaffin cell synapse includes the activation of nAChRs by the endogenously released ACh, depolarization of chromaffin cells by Na+ entry through nAChRs, the triggering of action potentials by Na+ entry through voltage-dependent Na+ channels, the entry of Ca2+ through VACCs, the increase of [Ca2+]c, and the activation of the exocytotic catecholamine release response (García et al., 2006; de Diego et al., 2008; Mahapatra et al., 2012). Thus, although duloxetine blocked IACh and the ACh-evoked [Ca2+]c transients and secretion responses, additional blockade of Na+ and/or Ca2+ channels could have contributed to those effects. This was tested in the present experiments.
Supplementary Fig. 1A shows a family of inward current traces obtained in a BCC voltage clamped at −80 mV and challenged with repeated 50-millisecond depolarizing test pulses to 0 mV applied at 20-second intervals. The initial fast inactivating peak current corresponds to INa and the slow inactivating near plateau current corresponds to ICa (arrows). INa and ICa were unaffected by 10 µM duloxetine, as shown in the superimposed traces of Supplementary Fig. 1B. Furthermore, in the time course graph of ICa peak displayed in Supplementary Fig. 1C, the increasing duloxetine concentrations did not significantly affect the initial current of 685 pA, that was fully suppressed by 100 µM Cd2+ (a VACCs blocker). Pooled data of current-voltage curves graphed in Supplementary Fig. 1 demonstrate that duloxetine had no significant effect either on ICa (Supplementary Fig. 1D) or INa (Supplementary Fig. 1E).
Effects of Duloxetine on K+-Elicited [Ca2+]c Transients and the Secretion of Catecholamine.
High K+ concentrations cause the direct depolarization of BCCs (Orozco et al., 2006) and the direct recruitment of VACCs with concomitant [Ca2+]c elevations and the triggering of secretion (Douglas and Rubin, 1963). Because duloxetine did not affect ICa, it was expected that the drug would not affect either the K+-elicited [Ca2+]c transients or the ensuing catecholamine secretion.
Supplementary Fig. 2A displays original [Ca2+]c traces obtained from a cell that was challenged with repeated K+ pulses (75 mM, low Na+, 75 K+; for 2 seconds, given at 2-minute intervals) The transient amplitudes were quite similar for all K+ pulses. Pooled data corroborate that duloxetine did not block those transients (Supplementary Fig. 2B).
The K+-elicited secretory responses matched those of [Ca2+]c transients. Thus, Supplementary Fig. 2C displays those responses in control conditions and Supplementary Fig. 2D displays the responses of cells exposed to 10 µM duloxetine. Pooled data indicated that at 1, 3, and 10 µM duloxetine the K+-evoked secretion responses overlapped with the control responses (Supplementary Fig. 2E).
Blockade by Duloxetine of Nicotinic Currents in Cell Lines Expressing Human α7, α3β4, or α4β2 nAChRs.
The whole-cell IACh of BCCs are plausibly carried by peripheral ganglionic-type nAChRs subunits α3β4 and α5 (Campos-Caro et al., 1997) and likely, also by α7 receptors (López et al., 1998; Fuentealba et al., 2004). The most abundant nAChRs in the brain, however, are known to be of the α7 and α4β2 subtypes (Dineley et al., 2015). It was therefore of interest to test whether duloxetine blockade of nicotinic currents carried by each receptor subtype separately expressed in cell lines, exhibited different potencies. This was achieved through voltage-clamp experiments performed with protocols similar to those of BCCs.
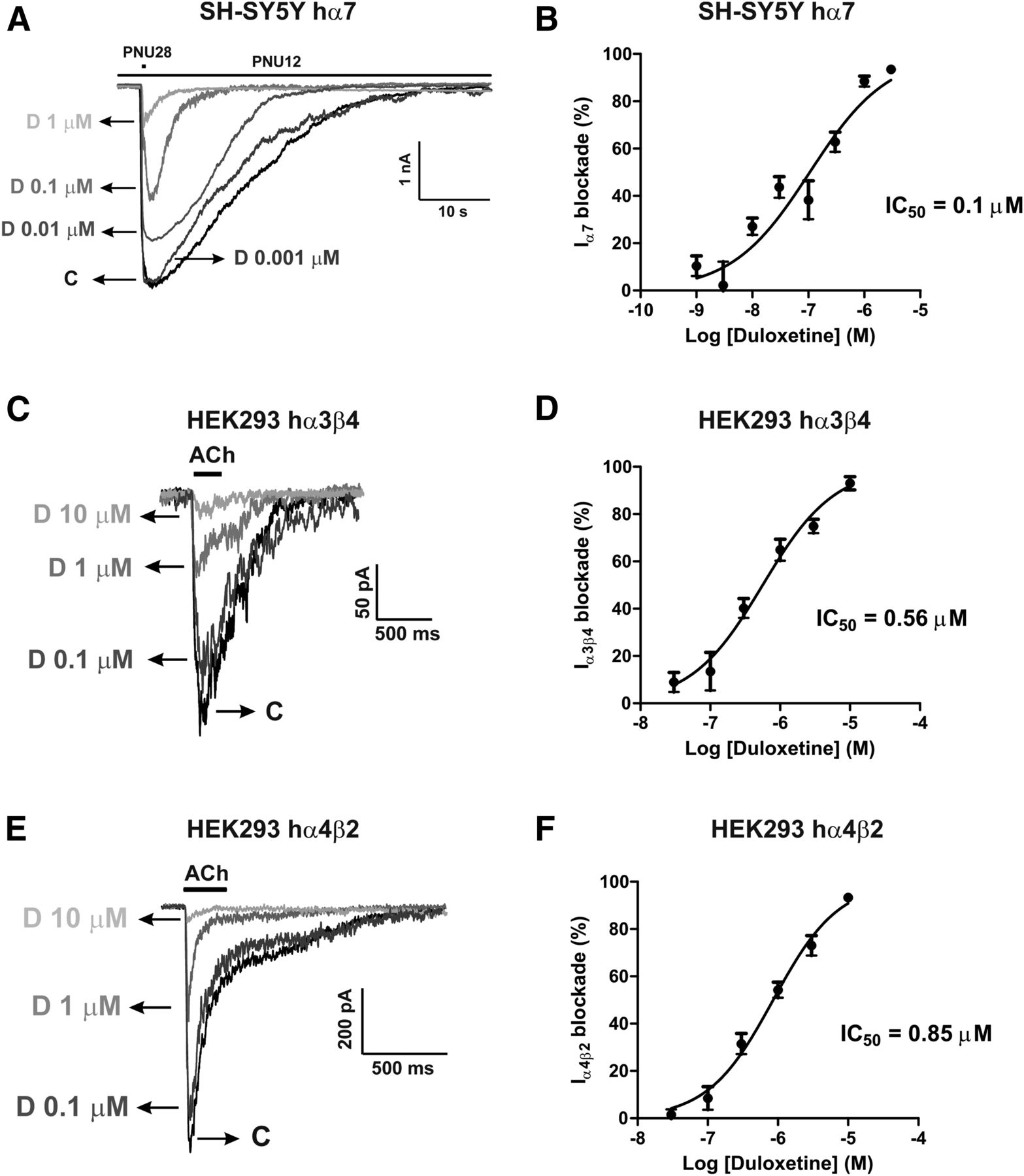
Example current traces obtained in SH-SY5Y cells overexpressing human α7 nAChRs and HEK293 cells expressing α3β4 and α4β2 receptors are drawn, respectively, in Fig. 7, A, C, and E. To estimate the blockade exerted by duloxetine on nicotinic currents data were normalized, within each individual cell, as the percentage of the initial current amplitude. Pooled data from 6 to 12 cells are plotted in Fig. 7, B, D, and F as concentration-effect sigmoid curves. The IC50 values for duloxetine blockade calculated from these curves were 0.1, 0.56, and 0.85 µM, respectively, for α7, α3β4, and α4β2 nAChRs. Thus, duloxetine blocked α7 receptors with potencies 5.6-fold and 8.5-fold higher than α3β4 and α4β2 receptors, respectively.

Blockade by duloxetine of nicotinic currents in cell lines expressing human α7, α3β4, or α4β2 nAChRs subtypes. (A, C, and E) Example current traces of α7, α3β4, or α4β2 nAChRs subtypes, respectively, subjected to different concentrations of duloxetine. Nicotinic current through α7 nAChRs was generated by 500-millisecond PNU282987 (10 µM) pulses every 60 seconds, in the presence of the nAChR allosteric modulator PNU120596 (10 µM), while nicotinic currents through α3β4 and α4β2 nAChRs were generated by 250-millisecond ACh (100 µM) pulses every 30 seconds. In each individual cell, the initial peak current amplitude (once the nicotinic current was stabilized) was normalized to 100% [control current (C)] and the successive currents obtained in the presence of the various concentrations of duloxetine (D) were expressed as the percentage of that initial nicotinic current. Normalized concentration-response curves for current inhibition are plotted in (B), (D), and (F), respectively, for the blockade by duloxetine of α7, α3β4, and α4β2 currents. IC50 values for such blockade are given in these panels. Data are the mean ± S.E.M. of 6–12 cells.
Discussion
The blockade of IACh exerted by duloxetine is the central finding in this study. Because the extent of blockade was similar at increasing ACh concentrations, and exhibited voltage and use dependence, it seems reasonable to conclude that such a blockade exhibits a noncompetitive nature. This resembles the mechanism underlying the blockade of nAChRs by the classic ganglionic blocking agent mecamylamine that in BCCs also exhibits voltage dependence (Nooney et al., 1992) and in rat chromaffin cells seems to penetrate and block the nAChR ion pore that would subsequently close and trap the antagonist inside the closed pore (Giniatullin et al., 2000).
Using mostly cell systems expressing various subtypes of nAChRs, various antidepressants have been shown to block those receptors. For instance, fluoxetine behaves as a reversible blocker of wild and mutant chick α7 receptors expressed in Xenopus oocytes (Maggi et al., 1998). In another rather extensive study (Fryer and Lukas, 1999a) that monitored 86Rb+ efflux in clonal cell lines, it was found that sertraline, fluoxetine, paroxetine, nefazodone, and venlafaxine behaved as functional noncompetitive blockers of nAChR subtypes α1β1γδ, α3β4 α5, and α7, with IC50 values in the range of 3–10 µM. It was remarkable that dual SNRI venlafaxine was the least potent in blocking 86Rb+ efflux stimulated by carbachol, in SH-SY5Y cells expressing α3β4α5 nAChRs, with an IC50 value of 12 µM. This relatively poor potency contrasts with our finding with duloxetine that inhibited IACh with an IC50 value of 0.86 µM in BCCs. The IC50 of duloxetine to block the native nAChRs of BCCs (Fig. 1D) is similar to the one we found in HEK293 cells expressing α4β2 receptors (0.85 µM) (Fig. 7F) and slightly higher than that found in HEK293 cells expressing α3β4 receptors (0.56 µM, Fig. 7D). Of interest was the finding that the IC50 of duloxetine blockade of α7-mediated nicotinic current in SH-SY5Y cells was substantially lower (0.1 µM) (Fig. 7B). This means that α7 nAChR blockade by duloxetine is exerted with about 5-fold to 10-fold higher potency with respect to currents carried by α3β4 or α4β2 receptors (this study) and about 30-fold to 100-fold more potent than several other antidepressants including dual venlafaxine, which is described above.
The physiologic secretion of adrenaline from adrenal medulla into the circulation under a stressful conflict is initiated by stimulation of nAChRs on the surface of chromaffin cells by endogenously released ACh. This causes an augmentation of cell excitability, the opening of voltage-activated sodium channels, the triggering of Na+-dependent action potentials, the recruitment of VACCs, the increase of Ca2+ entry, an augmented [Ca2+]c, and the exocytotic Ca2+-dependent release of adrenaline (Douglas, 1968; de Diego et al., 2008; Mahapatra et al., 2012). The blockade of nAChRs by duloxetine implied that all those subsequent events will also be blocked, as happened to be the case for cell excitability (Fig. 4), [Ca2+]c transients (Fig. 5), and catecholamine secretion (Fig. 6) in BCCs stimulated with ACh. However, the direct blockade of Na+ channels or VACCs by duloxetine could also lead to blockade of the subsequent [Ca2+]c transients and exocytotic responses. Because duloxetine did not affect INa or ICa (Supplementary Fig. 1), it was predictable that the K+-elicited [Ca2+]c transients and catecholamine release, which are mediated by Ca2+ entry through VACCs, will not be affected by the drug when stimulating the cells with a high K+ solution, as happened to be the case (Supplementary Fig. 2). Thus, duloxetine is eliciting a quite selective blockade of nAChRs of BCCs, without affecting sodium and calcium channels.
Now the question emerges of what subtypes of nAChRs is duloxetine acting upon. The bovine adrenal medullary chromaffin cells are known to express the autonomic type of α3β4α5 receptors (Criado et al., 1992; Campos-Caro et al., 1997) as well as functionally defined α7 nAChRs (López et al., 1998; Fuentealba et al., 2004). Thus, we may conclude that duloxetine blocks those peripherally expressed nAChR subtypes. Three facts indicate that duloxetine may also target the brain most abundant α7 and α4β2 receptors: 1) antidepressants such as imipramine (Eldefrawi et al., 1981; Arita et al., 1987), amitriptyline and nortriptyline (Schofield et al., 1981), desipramine (Rana et al., 1993), maprotiline (Arita et al., 1987), and fluoxetine (García-Colunga et al., 1997; Hennings et al., 1997; Maggi et al., 1998) exhibit a high degree of promiscuity in blocking the different subtypes of peripheral and central nAChRs; 2) the central and peripheral α7 nAChRs exhibit similar structural, electrophysiological, and pharmacological properties (Criado et al., 1992; Campos-Caro et al., 1997); and 3) in SH-SY5Y overexpressing human α7 receptors, we have found that duloxetine inhibited nicotinic currents with an IC50 of 0.1 µM (Fig. 7B). Similarly, in HEK293-hRα3β4 and HEK293-hRα4β2, we have found that duloxetine inhibited nicotinic currents with IC50 values of 0.56 and 0.85 µM, respectively (Fig. 7, D and F).
The interesting hypothesis that cholinergic hyperactivity is involved in the pathogenesis of depression (Janowsky et al., 1972) is concordant with studies showing a correlation of smoking and nicotine consumption with depression and mood disorders (Anda et al., 1990; Glassman et al., 1990), and with the proposal that nAChR blockade could be therapeutically exploited to design novel clinical useful antidepressants (Fryer and Lukas, 1999b). A major challenge to the hypothesis is the pharmacokinetic profile of most antidepressants, whose calculated brain concentrations reached in patients is in the nanomolar range (Weber et al., 2013). This is far away from the calculated IC50 values to block the various subtypes of nAChRs on in vitro systems, namely 2–8 μM for fluoxetine, paroxetine, and sertraline to block α4β2 receptors, 2.6–11 µM to block α3β4 receptors, and 2–20 µM to block α7 receptors (Fryer and Lukas, 1999a; Arias et al., 2010b). In clear contrast with these values are those reported for mecamylamine, namely 0.09–1.18 µM to block α3β4 receptors, 0.44–0.62 µM to block α3β4α5 receptors, and 0.6–3 µM to block α4β2 receptors (Giniatullin et al., 2000; Papke et al., 2001.) In our present study, of particular relevance is the fact duloxetine blocked the α7 nAChR currents with an IC50 of only 100 nM; these concentrations are likely in the range of those achieved by duloxetine in depressed patients, as a maximum plasma concentration of approximately 150–370 nM has been described during duloxetine treatment (Knadler et al., 2011).
Although the classic ganglionic blocking agent mecamylamine cannot be included in the classes of antidepressant drugs cited above, it however exhibits mood-improving activity (Shytle et al., 2002). It is interesting that duloxetine closely mimics the properties of mecamylamine in blocking the nAChRs, namely, their noncompetitive nature, and their IC50 value of 0.86 µM to block IACh (Fig. 1) and ACh-triggered exocytosis (Fig. 6). It is also interesting that in populations of BCCs the duloxetine-elicited blockade of IACh and secretion develops slowly, indicating its possible membrane accumulation in the lipid bilayer close to nAChRs, thus serving as a kind of reservoir for the gradual delivery of the drug into the nAChR neighborhood. This slow steady-state equilibrium is typical of other drugs that block VACCs of BCCs, such as flunarizine, which has characteristics of a pseudoirreversible blockade (Novalbos et al., 1999). All of these characteristics lead to the conclusion that duloxetine may reach clinically relevant brain concentrations in the nanomolar range to cause at least some blockade of nAChRs, particularly the α7 nAChRs that are inhibited with an IC50 of 0.1 µM, which is 10-fold to 100-fold lower than the IC50 values reported for nAChR blockade by several other antidepressants. Although this proposal may have some support in the findings of this study, it should be tempered by the fact that we explored the blocking effects of duloxetine on α7 PNU120596-potentitated currents. We had to take advantage of this larger current because ACh elicited only a tiny current when applied to SH-SY5Y neuroblastoma cells overexpressing α7 nAChRs (data not shown). However, it has previously been shown that PNU120596-potentiated currents have different pharmacological sensitivity to noncompetitive antagonists that block channels activated by ACh in the absence of the positive allosteric modulator. For instance, the classic noncompetitive nAChR blocker mecamylamine showed different IC50 values to block control and PNU120596-modified receptors (Peng et al., 2013).
In conclusion, we have found here that the dual SNRI antidepressant duloxetine inhibits with high potency (0.1–0.86 µM) the nicotinic currents of the α7, α3β4, and α4β2 nAChR subtypes. In so doing, duloxetine also blocks the functions mediated by such receptors when stimulated by ACh, namely, enhanced cell excitability, [Ca2+]c transients, and catecholamine release in bovine adrenal medullary chromaffin cells. This may contribute to the antidepressant and/or side effects of duloxetine in clinical settings.
Acknowledgments
We thank Novartis Pharma AG (Switzerland) for providing the cell lines overexpressing human α7 (SH-SY5Y-hRα7), α3β4 (HEK293-hRα3β4), and α4β2 (HEK293-hRα4β2). We also thank Fundación Teófilo Hernando for continued support.
Authorship Contributions
Participated in research design: Padín-Nogueira, Gandía, and García.
Conducted experiments: Nanclares, Gameiro-Ros, Méndez-López, Martínez-Ramírez, Padín-Nogueira, Colmena, and Baraibar.
Performed data analysis: Nanclares, Gameiro-Ros, Méndez-López, Martínez-Ramírez, Colmena, and Baraibar.
Wrote or contributed to the writing of the manuscript: Nanclares, Padín-Nogueira, Gandía, and García.
Footnotes
- Received May 28, 2018.
- Accepted July 12, 2018.
↵1 L.G. and A.G.G. are co-senior authors and contributed equally to this work.
This work was supported by grants from the Spanish Ministerio de Economía y Competitividad [SAF2013-44108-P and SAF2016-78892-R] to A.G.G. and L.G., and a grant from Fundación Eugenio Rodriguez Pascual to Luis Gandía.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ACh
- Acetylcholine
- BCC
- bovine chromaffin cell
- [Ca2+]c
- cytosolic calcium
- DMEM
- Dulbecco’s modified Eagle’s medium
- FBS
- fetal bovine serum
- Fura-2/AM
- Fura-2/acetoxymethyl ester
- HP
- holding potential
- 5-HT
- serotonin
- IACh
- inward current through nicotinic receptors
- ICa
- inward current through voltage-activated calcium channels
- INa
- inward current through voltage-activated sodium channels
- NA
- noradrenaline
- nAChR
- nicotinic acetylcholine receptor
- SNRI
- serotonin and noradrenaline reuptake inhibitor
- VACC
- voltage-activated calcium channel
- Vm
- membrane potential
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}