


Abstract
Phosphatidylinositol 3-kinase delta (PI3Kδ) is a critical signaling molecule in B cells and is considered a target for development of therapies against various B cell malignancies. INCB040093 is a novel PI3Kδ small-molecule inhibitor and has demonstrated promising efficacy in patients with Hodgkin’s lymphoma in clinical studies. In this study, we disclose the chemical structure and the preclinical activity of the compound. In biochemical assays, INCB040093 potently inhibits the PI3Kδ kinase, with 74- to >900-fold selectivity against other PI3K family members. In vitro and ex vivo studies using primary B cells, cell lines from B cell malignancies, and human whole blood show that INCB040093 inhibits PI3Kδ-mediated functions, including cell signaling and proliferation. INCB040093 has no significant effect on the growth of nonlymphoid cell lines and was less potent in assays that measure human T and natural killer cell proliferation and neutrophil and monocyte functions, suggesting that the impact of INCB040093 on the human immune system will likely be restricted to B cells. INCB040093 inhibits the production of macrophage-inflammatory protein-1β (MIP-1beta) and tumor necrosis factor-β (TNF-beta) from a B cell line, suggesting a potential effect on the tumor microenvironment. In vivo, INCB040093 demonstrates single-agent activity in inhibiting tumor growth and potentiates the antitumor growth effect of the clinically relevant chemotherapeutic agent, bendamustine, in the Pfeiffer cell xenograft model of non-Hodgkin’s lymphoma. INCB040093 has a favorable exposure profile in rats and an acceptable safety margin in rats and dogs. Taken together, data presented in this report support the potential utility of orally administered INCB040093 in the treatment of B cell malignancies.
Introduction
Lymphoid malignancies are among the leading causes of cancer death in the United States with 81,000 estimated new cases annually (Siegel et al., 2016). Among these malignancies, the majority are derived from B cells. Although the B cell–specific monoclonal anti-CD20 antibody rituximab and small-molecule Bruton’s tyrosine kinase inhibitor ibrutinib have significantly improved the prognosis for these patients, the annual mortality rate remains high with 12,000 estimated deaths in 2016.
The recognition that aberrant signal transduction occurs in malignant cells and is involved in tumor progression has led to a focus on agents that modulate signaling pathways. Phosphatidylinositol 3-kinase (PI3K) belongs to a family of lipid signaling kinases that phosphorylate phosphoinositides at the D3 position of the inositol ring (Cantley, 2002). PI3Ks are divided into three classes (class I, II, and III) according to their structure, regulation, and substrate specificity. Class I PI3Ks, which include four isoforms (α, β, γ, and δ), are dual-specificity lipid and protein kinases that catalyze the phosphorylation of phosphatidylinositol-4,5-bisphophate, giving rise to phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 functions as a second messenger that controls a number of cellular processes, including growth, survival, adhesion, and migration, by serving as a docking site for plasma membrane recruitment of many pleckstrin homology domain-containing proteins, such as protein kinase B/Akt (Okkenhaug and Vanhaesebroeck, 2003).
The δ isoform of the class 1 PI3K (PI3Kδ) has been shown to be critical for B cell signaling and function (Fung-Leung, 2011). It is expressed exclusively in immune cells and preferentially in B cells (Okkenhaug et al., 2007). Mice deficient in PI3Kδ function or protein, although fertile and have a normal life span, demonstrate specific defects in B cell signaling that lead to impaired B cell development and reduced antibody production after antigen stimulation (Jou et al., 2002; Okkenhaug et al., 2002). B cells derived from PI3Kδ-deficient mice or wild-type B cells treated with PI3Kδ inhibitor all have reduced proliferative responses to anti-IgM, anti-CD40, or interleukin (IL)-4 stimulation (Clayton et al., 2002; Bilancio et al., 2006).
Although no mutations have been identified in PI3Kδ in B cell malignancies, forced expression of PI3Kδ was shown to be transforming in cell lines (Kang et al., 2005), and consistent overexpression of PI3Kδ has been observed in acute myeloblastic leukemia (Sujobert et al., 2005). Given the unique expression and pivotal function of PI3Kδ in B cell growth and development, one could speculate that unregulated PI3Kδ activity could result in B cell proliferative diseases. In fact, CAL-101 (idelalisib), a selective PI3Kδ inhibitor, has been shown to inhibit PI3K signaling and B cell tumor growth in preclinical studies (Herman et al., 2010; Lannutti et al., 2011; Meadows et al., 2012) and was approved for the treatment of patients with relapsed follicular B cell non-Hodgkin lymphoma and relapsed chronic lymphocytic leukemia (Brown et al., 2014; Fruman and Cantley, 2014; Gopal et al., 2014). The PI3Kδ selective inhibitor TGR-1202 and the δ isoform-biased PI3Kδ/γ dual-inhibitor IPI-145 (duvelisib) are also in late-stage clinical studies for the similar indications. These results indicate that PI3Kδ is an attractive target for drug development against various B cell malignancies.
INCB040093 is a novel, selective, and potent small-molecule inhibitor of PI3Kδ that is being tested in clinical studies in patients with B cell malignancies. Interim analysis of a phase I study shows that in heavily pretreated patients with refractory or relapsed classic Hodgkin lymphoma INCB040093 at 100 mg twice daily monotherapy generated an overall response rate of 36% and disease control rate of 55% (Phillips et al., 2015). Furthermore, in combination with the Janus kinase (JAK)1-selective inhibitor itacitinib, the overall response rate and disease control rate were 65% and 80%, respectively, and several patients experienced a complete response (Phillips et al., 2015). To corroborate with the clinical study results, we disclose in this report the chemical structure of INCB040093 and the preclinical profile of this compound, including activities in biochemical, cellular, and whole-blood assays for PI3Kδ; antitumor efficacy in B cell tumor models; pharmacokinetic properties; and safety profile. These preclinical study results fully support INCB040093 as a potential therapeutic agent for treating B cell malignancies.
Materials and Methods
Enzyme Assays.
The PI3K assays were carried out at room temperature in 20 mM 4-morpholinepropanesulfonic acid, pH 6.7, 10 mM MgCl2, 5 mM dithiothreitol, and 0.03% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid in 96-well plate in a final volume of 24 μl. The reaction mixture contained 50 μM D-myo-phosphatidylinositol 4,5-bisphosphate (Echelon Biosciences, Salt Lake City, UT), the kinase, and varying concentration of INCB040093, initiated by the addition of ATP to a final concentration of 1000 μM containing 2.2 μCi [γ-33P]ATP (Perkin-Elmer, Boston, MA), and terminated by the addition of 100 μl quench buffer (1 M potassium phosphate, pH 8.0, 30 mM EDTA) after incubation for 180 minutes. The reaction solution was then filtered with 96-well MultiScreen IP 0.45 μm polyvinylidene difluoride filter plates (Millipore, Billerica, MA). The filter plate was washed with 18 × 200 μl wash buffer containing 1 M potassium phosphate (pH 8.0) and 1 mM ATP. The plate was air dried at 37°C overnight. The radioactivity of the product was determined by scintillation counting on Topcount (Perkin-Elmer). IC50 determination was performed by curve fitting using the GraphPad Prism 3.0 software.
The kinase-profiling assays were carried out at 1 mM ATP using homogeneous time-resolved fluorescence assay format, according to manufacturer’s instruction, provided by Cisbio US (Bedford, MA). A total of 57 kinases was tested, including AKT1, AURKA, AXL, BRAF, BTK, CAMK2D, CDK2, CHEK1, CHEK2, CLK3, DDR2, DYRK2, EGFR, EPHA2, EPHB4, ERBB4, FAK, FGFR3, FLT3, GSK3B, HIPK3, IGF1R, IKKA, IKKB, JNK1, KDR, KIT, LCK, LOK, LRRK2, MAP3K8, MAPKAPK2, MINK, MSSK1, NLK, PAK4, PDGFR BETA, PHKG2, PIM1, PIM2, PKCD, PLK3, PRKAA1, RET, ROCK1, RPS6KB1, RSK2, SGK1, SGK2, SGK3, SRC, STK4, SYK, TGFBR1, TIE2, TRKA, and ZAP70 (Millipore).
Primary Immune Cell Proliferation Assays.
For the dog B cell proliferation assay, fresh dog blood was separated by standard density gradient centrifugation on Ficoll-Hypaque (GE Healthcare, Uppsala, Sweden). The resulting peripheral blood mononuclear cells (PBMCs) were cultured in RPMI 1640 + 10% fetal bovine serum (FBS) in a flask overnight to let monocytes adhere. On the next day, the floating cells were collected, centrifuged, and resuspended in red blood cell (RBC) lysis buffer (eBiosciences, San Diego, CA) for 10 minutes on ice. The remaining cells were washed with phosphate-buffered saline (PBS) twice and resuspended in PBS + 2 mM EDTA + 0.5% bovine serum albumin to a final density of 1 × 108 cells in 0.5 ml. The cells were then incubated with mouse anti-canine CD21 antibody (Serotec, Raleigh, NC) for 15 minutes at 4°C and then washed with PBS + 2 mM EDTA twice. The CD21+ cells were then purified with rat anti-mouse IgG1 (Miltenyi Biotec, San Diego, CA). To measure the antiproliferation activity of INCB040093 against dog B cells, the canine CD21+ cells (2 × 105 cells/well per 200 μl) in RPMI 1640 + 10% FBS with or without a wide range of concentration of INCB040093 were stimulated with goat anti-dog IgM (15–30 μg/ml) (KPL (Gaithersburg, MD) or Serotec) for 3 days before [3H]thymidine (1 µCi/well) (Perkin-Elmer) was added. After overnight culture, the incorporated radioactivities were measured by liquid scintillation counting with a TopCount (Packard Bioscience, Meridan, CT).
To acquire human B, T, and natural killer (NK) cells, human PBMCs were isolated from the peripheral blood of normal and drug-free donors by standard density gradient centrifugation on Ficoll-Hypaque (GE Healthcare) and incubated with anti-CD19, anti-CD3, and anti-CD56 microbeads (Miltenyi Biotec), respectively. The primary cells were then purified by positive immunosorting using an autoMacs (Miltenyi Biotec), according to the manufacturer’s instructions. The fraction 2000 from elutriation of human PBMC was also collected and considered as total T cell population. In proliferation assays, purified CD19+ human B cells (2 × 105/well per 200 µl) were cultured in 96-well ultra-low binding plates (Corning, Corning, NY) in RPMI 1640 + 10% FBS with a wide range of concentration of INCB040093 and stimulated for 3 days with goat F(ab′)2 anti-human IgM (15 µg/ml; Invitrogen, Carlsbad, CA), IL-4 (20 ng/ml), lipopolysaccharide (LPS; 1 μg/ml), soluble CD40 ligand (1 μg/ml), anti-CD40 antibody (15 μg/ml), or B cell activating factor (BAFF; 10 ng/ml) (all from R&D Systems, Minneapolis, MN). Purified CD56+ NK cells (2 × 105/well per 200 µl) were stimulated for 3 days with IL-2 (5 ng/ml; R&D Systems) in the presence of a wide range of concentration of INCB040093. Purified CD3+ T cells and total T cell population (elutriation fraction 2000) (2 × 105/well per 200 µl) were treated with a wide range of concentration of INCB040093 and stimulated for 3 days with IL-2 (5 ng/ml; R&D Systems) and anti-CD3 antibody (100 ng/ml; eBiosciences) + anti-CD28 antibody (R&D Systems), respectively. [3H]Thymidine (1 µCi/well) (PerkinElmer) in PBS was then added to the cell culture for additional 12–16 hours before the incorporated radioactivity was separated by filtration with water through GF/B filters (Packard Bioscience) and measured by liquid scintillation counting with a TopCount (Packard Bioscience).
Lymphoma Cell Line Proliferation, Viability, and Apoptosis Assays.
Pfeiffer, SU-DHL-6, Rec-1, Mino, Jeko-1, and HS-445 cell lines were purchased from American Type Culture Collection (Manassas, VA). SU-DHL-1, SU-DHL-4, SU-DHL-5, SU-DHL-8, SU-DHL-10, SU-DHL-16, WSU-NHL, and U-HO1 cell lines were obtained from DSMZ (Braunschweig, Germany). All the cell lines were maintained according to the recommendations of suppliers. To measure the antiproliferation activities of INCB040093 against these tumor cell lines, the cells were plated in their respective culture medium (2 × 103 cells/200 μl per well) into 96-well ultralow binding plates (Corning), with or without a wide range of concentration of INCB040093. After 3–4 days, [3H]thymidine (1 µCi/well; PerkinElmer) in PBS was then added to the cell culture for an additional 12–16 hours before the incorporated radioactivity was separated by filtration with water through GF/B filters (Packard Bioscience) and measured by liquid scintillation counting with a TopCount (Packard Bioscience).
Cell viability assay was conducted using the cell titer glow (Promega, Madison, WI), according to manufacturer’s instruction, in the presence or absence of INCB040093.
For apoptosis assay, cells were cultured in the presence or absence of INCB040093 (1 μM) for 24 hours. The percentage of apoptotic cells was determined by annexin V-fluorescein isothiocyanate/propidium iodide (eBioscience) staining, followed by two-color flow cytometry analysis. The percentage data represent both annexin V single-positive and annexin V/propidium iodide double-positive cells.
Cytokine and Chemokine Production Assays.
To evaluate IL-17 production from human T cells, human T helper (Th)17 cells were generated, as described previously, with some modifications (Volpe et al., 2008). In brief, human naive CD4+CD45RA+ T cells were purified from PBMCs using the Naive T Cell Isolation kit (Miltenyi Biotec), resuspended in culture medium (RPMI 1640 + 10% FBS), and seeded in 24-well plates (1 × 106/ml/well). The cells were cultured for up to 7 days at 37°C in an atmosphere of 5% CO2, without any media change, in the presence of a wide range of INCB040093 plus IL-1β (20 ng/ml), IL-6 (30 ng/ml), IL-23 (30 ng/ml), transforming growth factor-β (2.25 ng/ml), anti–interferon-γ (1 μg/ml) antibody, anti–IL-4 (2.5 μg/ml) antibody (R&D Systems), and microbeads coated with anti-CD2, anti-CD3, and anti-CD28 antibodies (Milenyi Biotec). The bead to cell ratio ranged from 1:1 to 3:1. The cell culture supernatants were collected and measured for their content of IL-17 by enzyme-linked immunosorbent assay (R&D systems), according to the manufacturer’s protocol.
To measure PBMC tumor necrosis factor (TNF)-α production, PBMCs were isolated by standard density gradient centrifugation on Ficoll-Hypaque (GE Healthcare). The cells were plated in 250 μl RPMI 1640 at 2 × 106/ml in 96-well plates in the presence of a wide range of INCB040093 for 10 minutes at 37°C. LPS at 1 μg/ml was then added for 5 hours at 37°C. Supernatants were collected, and the TNF-α levels were measured with human TNF-α Quantikine enzyme-linked immunosorbent assay kits (R&D Systems), according to manufacturer’s instruction.
The levels of secreted cytokines and chemokines in the supernatants of the coculture between HS-445 [Hodgkin’s lymphoma (HL) cell line] and HS-5 (stroma cell line) were determined using a Luminex assay specific for a broad panel of human cytokines and chemokines (EMD Millipore, Burlington, MA). The cells were cocultured at different ratios for 3 days in the presence or absence of INCB040093. Standard curves for each cytokine and chemokine were run, and the concentrations in the samples were calculated using Exponent 3.1 software (Luminex, Austin, TX).
Human Neutrophil Elastase Exocytosis Assay.
Human neutrophil elastase exocytosis assay was conducted, as described previously, with some modifications (Sadhu et al., 2003). In brief, fresh human blood was separated by standard density gradient centrifugation on Ficoll-Hypaque (GE Healthcare). The resulting cell pellet was treated with RBC lysis buffer for 5 minutes at room temperature. After washing with PBS, the remaining cells were considered as purified neutrophils. The purified neutrophils with or without a wide range of INCB040093 were stimulated with N-formyl peptide (fMLP; 1 μM) in PBS containing cytochalasin B (10 μg/ml), sodium azide (1 μM), and L-methionine (5 μg/ml) (Sigma-Aldrich, St. Louis, MO) for 60 minutes at 37°C in a 96-well culture plate coated with fibrinogen (2 mg/ml; Sigma-Aldrich). At the end of the incubation, the plate was centrifuged for 5 minutes, and 90 μl supernatant was transferred to wells containing 10 μl elastase substrate solution (MeO-suc-Ala-Ala-Pro-Val-pNA) (10 mM; Sigma-Aldrich). Absorbance at 410 minutes was monitored for up to 30 minutes in a 96-well plate reader.
Chemotaxis Assay.
Human monocytes were isolated from PBMC via centrifugal elutriation and allowed to rest overnight at 4°C. The rested cells were washed and resuspended in serum-free media. Media with or without CC chemokine ligand (CCL)2 (10 nM; R&D Systems) in the presence or absence of INCB040093 was added to the bottom wells of a prewarmed 96-well Boyden chemotaxis chamber. An 8-μm filter was placed on the top, and the lid was closed. Monocytes (4 × 105) were then added to the holes in the chamber lid, which is associated with the chamber well below the filter membrane. A plate sealer was placed across the top, and the whole chamber unit was incubated at 37°C, 5% CO2, for 45 minutes. After the cells were aspirated off, the chamber lid was opened, and the filter was gently removed. The top of the filter was washed three times with PBS, and the bottom was left untouched. The filter was air dried and stained with Wright Giemsa stain. The migrated cells on the bottom of the filter were counted by light microscopy.
Induced and Constitutive Signaling Assays.
PI3Kα, β, γ, and δ isoform-specific signaling was determined by measuring the level of pAkt (Ser473) in NIH3T3, RAW264.7, PC-3, and Ramos cells (American Type Culture Collection) after stimulation with platelet-derived growth factor (PDGF), lysophosphatidic acid (LPA), C5a, and anti-IgM antibody, respectively. Specifically, NIH3T3, PC-3, RAW264.7, and Ramos cells were cultured in the American Type Culture Collection–recommended medium in six-well plates for 24 hours, serum starved overnight, incubated with a wide range of INCB040093 for 2 hours, and then treated with PDGF BB (20 ng/ml), LPA (10 nM), C5a (50 nM) (R&D Systems), and anti-IgM (4 μg/ml) (Invitrogen), respectively, for 15–30 minutes at 37°C. After stimulation, the cells were pelleted and lysed with lyisis buffer (Cell Signaling, Danvers, MA). The lysates were separated by SDS-PAGE (4%–12%) and transferred to nitrocellulose membrane. The blots were then probed with an anti-pAkt (Ser473) antibody (Cell Signaling). The bound antibodies were detected by chemiluminescence using SuperSignal West Dura Luminol/Enhancer Solution (Thermo Fisher Scientific, Waltham, MA). The membranes were then stripped and reprobed with an anti-Akt antibody (Cell Signaling) for demonstration of loading control.
To examine the effect of PI3Kδ inhibition on constitutive PI3K signaling in hematologic tumor cells, the Pfeiffer cells were incubated with different amounts of INCB040093 for 3–4 hours at 37°C in culture medium, collected by centrifugation, and lysed with 1× lysis buffer (Cell Signaling). The cell lysates were further sonicated briefly and incubated on ice for 30 minutes before supernatants were collected, separated by SDS-PAGE (4%–12%), and transferred to nitrocellulose membrane with an iBlot device (Invitrogen). The blots were probed with an anti-pAkt (Ser473; Cell Signaling). Nitrocellulouse-bound antibodies were detected by chemiluminescence using SuperSignal West Dura Luminol/Enhancer Solution (Thermo Fisher Scientific). The membranes were then stripped and reprobed with an anti-Foxo3a (Thr32; Cell Signaling) antibody for further downstream signaling and reprobed again with an anti-Akt antibody (Cell Signaling) to detect total Akt protein.
To measure signal transducer and activator of transcription (STAT)3 phosphorylation in various lymphoma cell lines, the cell assays were cultured in the presence of HS-5 bone marrow stroma cell conditioned medium, IL-6 (10 ng/ml), or IL-10 (10 ng/ml) for 3 days. The cell lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane with an iBlot device (Invitrogen). The blots were probed with an anti-pSTAT3 (Y705) antibody (Cell Signaling). Nitrocellulose-bound antibodies were detected by chemiluminescence using SuperSignal West Dura Luminol/Enhancer Solution (Thermo Fisher Scientific).
Human Whole-Blood Assays.
The effect of PI3Kδ inhibition on anti-IgE antibody-induced CD63 expression on basophils in human whole blood was evaluated using the Flow Cast kit (ALPCO, Salem, NH), according to the manufacturer’s instructions.
The effect of PI3Kδ inhibition on constitutive pAkt in tumor cells in human whole blood was measured by spiking Pfeiffer cells (5 × 105 in 5 μl RPMI 1640 + 10% FBS) into 25 μl fresh human blood (25 μl), incubated with a wide range of INCB040093 at 37°C for 2 hours, and then fixed with 3% formaldehyde. After lysis of RBC with 1× RBC lysis buffer (eBioscience), the remaining cells were permeabilized with 50% methanol in PBS and resuspended in 100 μl PBS + 0.5% BSA before incubation with an Alexa-488–conjugated anti-pAkt (Ser478) antibody (Cell Signaling) for 1 hour at room temperature. After washing with PBS, the percentage of Pfeiffer cells positive for pAkt was analyzed by fluorescence-activated cell sorting.
Pharmacokinetic Studies.
Male rats were used in this study. Rats were given either a single 5 mg/kg intravenous bolus injection (IV) (N = 3) or a single 10 mg/kg oral dose (PO) (N = 3) of INCB040093. All doses and concentrations are expressed as free base equivalent. The vehicle for i.v. dosing was 5% dimethylacetamide (DMAC) and 10% of propylene glycol in saline. The vehicle for PO dosing was 5% DMAC in 0.5% methylcellulose aqueous solution. Blood samples were collected at predose, 5 (for i.v. dosing only), 15, and 30 minutes, and 1, 2, 4, 6, 8, and 24 hours postdose in both i.v. and PO groups. All blood samples were collected using EDTA as the anticoagulant and centrifuged to obtain plasma samples. Urine samples were also collected and pooled (0–8 and 8–24 hours) to determine the extent of urinary excretion. Plasma and urine concentrations of INCB040093 were determined under non-GLP (good laboratory practice) condition utilizing a protein precipitation bioanalytical method. The calibration range was from a lower limit of quantitation of 0.002 μM to an upper limit of quantitation of 10 μM. The plasma concentration–time data were used to determine the pharmacokinetic parameters for each rat by standard noncompartmental methods using WinNonlin version 5.0.1 (Pharsight, Saint Louis, MO).
Xenograft Model.
Animals were housed in a barrier facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. All of the procedures were conducted in accordance with the US Public Service Policy on Human Care and Use of Laboratory Animals and with Incyte Animal Care and Use Committee Guidelines.
Xenograft studies were performed using 5- to 8-week-old female CB.17 SCID mice (Charles River, Wilmington, MA). Pfeiffer tumor fragments (approximately 3 × 3 mm) were implanted s.c. Tumors were measured after becoming visible two to four times weekly in two dimensions using a caliper, and the volumes were calculated using the following formula: V = 0.5(A × B2), where A and B are the long and short diameters of the tumor, respectively. Tumor-bearing animals were sorted into groups of six to eight with similar mean tumor volumes prior to treatment—usually 100–200 mm3. For all studies, INCB040093 was resuspended in 0.5% methocellulose (with or without the addition of 5% DMAC), and twice-daily dosing was administered at 8- and 16-hour intervals (specifically, at approximately 7 AM and 3 PM daily). Tumor growth inhibition, expressed in percentages, is calculated using the following formula: [1 − (volume [treated]/volume [vehicle])] × 100.
Results
INCB040093 Is a PI3Kδ Selective Inhibitor.
The biochemical potency of INCB040093 [(S)-7-(1-(9H-purin-6-ylamino)ethyl)-6-(3-fluorophenyl)-3-methyl-5H-thiazolo[3,2-α]pyrimidin-5-one] (Fig. 1) for the inhibition of human PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ was determined in enzymatic assays using recombinant truncated proteins for each of the class 1 PI3K family members. Phosphorylation of the lipid kinase substrate D-myo-phosphatidylinositol 4,5-bisphosphate was measured. All assays were conducted at 1 mM ATP, a concentration that is in the range of cellular ATP levels. The average IC50 values from replicate assays using multiple lots of INCB040093 are indicated in Fig. 1. Based on these data, INCB040093 potently inhibits PI3Kδ enzyme activity (IC50 = 31 nM), with 74- to >900-fold selectivity for the other PI3K family members. Idelalisib demonstrated similar activity as INCB040093 in the same assays (Fig. 1). Additional kinetic analyses determined that INCB040093 is a reversible and ATP competitive inhibitor of PI3Kδ (data not shown). Using similar assay conditions as the PI3K biochemical assays (i.e., 1 mM ATP), INCB040093 was selective against a panel of 57 additional kinases when tested at a concentration of 20 μM. In addition, INCB040093 did not significantly inhibit (<30% inhibition) a broad panel of 196 other kinases in two non-GLP Cerep kinase assay screens when tested at 1 μM (data not shown).

Chemical structure of INCB040093 [(S)-7-(1-(9H-purin-6-ylamino)ethyl)-6-(3-fluorophenyl)-3-methyl-5H-thiazolo[3,2-α]pyrimidin-5-one] and enzyme-inhibitory activity of INCB040093 against different members of the human class I PI3K family.
The activity and selectivity of INCB040093 against each one of the class I PI3K isoforms were further studied in signaling assays. Induction of pAkt by PDGF in NIH3T3 fibroblast cells, LPA in PC-3 prostate cancer cells, anti-IgM in Ramos Burkitt’s lymphoma cells, and C5a in RAW264.7 mouse macrophage cells was used to represent PI3Kα-, PI3Kβ-, PI3Kδ-, and PI3Kγ-mediated signaling, respectively. As shown in Fig. 2, whereas INCB040093 inhibited anti-IgM–induced pAkt in Ramos cells with an IC50 value of ∼10 nM, it did not significantly reduce the level of C5a-induced pAkt in RAW264.7 cells at concentrations up to 300 nM. In addition, concentrations of INCB040093 up to 3000 nM showed no activity in reducing the level of pAkt induced by PDGF in NIH3T3 fibroblasts and LPA in PC-3 cells. Thus, the results from both biochemical and signaling assays suggest that INCB040093 is a potent and selective PI3Kδ inhibitor.

INCB040093 is selective in inhibiting signaling by PI3K isoforms in cells. pAkt was induced by PDGF, LPA, C5a, and anti-IgM in NIH3T3, PC-3, RAW264.7, and Ramos cells, respectively, for PI3Kα-, PI3Kβ-, PI3Kγ-, and PI3Kδ-mediated signaling. The levels of pAkt and total Akt in the cell lysates were measured by Western blotting using an anti-pAkt (Ser473) and an anti-Akt antibody, respectively.
INCB040093 Inhibits PI3Kδ Functions in Primary Immune Cells.
PI3Kδ is known to be critical for B cell survival and proliferation by functioning downstream of B cell antigen receptor (BCR), prosurvival receptors such as BAFF receptor and the IL-4 receptor, costimulatory receptors such as CD40, and Toll-like receptors (Puri and Gold, 2012). PI3Kδ is also known to mediate Fcε receptor signaling-induced basophil activation (CD63 expression) (Lannutti et al., 2011) and has been shown to promote nuclear translocation of the transcriptional factor RORγ for the differentiation of naive T cell into Th17 phenotype (Kurebayashi et al., 2012). To evaluate the activity of INCB040093 against PI3Kδ functions in different immune cells, the effect of the compound on the proliferation of CD19+ human or CD21+ canine primary B cells induced by anti-IgM antibody, LPS, IL-4, IL-6, BAFF, anti-CD40, or soluble CD40 ligand was measured. After incubation for 3 days, INCB040093 potently inhibited the cellular proliferation ([3H]thymidine uptake) with these stimulants, with IC50 values ranging from 1 to 3 nM (Table 1). Similar activity was also found for INCB040093 in blocking human Th17 cell differentiation (Table 1). In addition, INCB040093 was very active in reducing anti-FcεR–induced basophil activation in the presence of human whole blood (IC50 = 33 nM) (Table 1). These results indicate that INCB040093 is active in inhibiting previously reported PI3Kδ functions in immune cells, further validating INCB040093 as a PI3Kδ inhibitor.
Inhibitory activity of INCB040093 against human primary immune cell
To evaluate the potential therapeutic utility of INCB040093, it was of interest to determine whether the compound impacts the functions of other immune cells in addition to those previously studied. As shown in Table 1, INCB040093 is approximately 1000-fold less potent in inhibiting human T cell proliferation mediated by T cell antigen receptor (TCR) activation with an anti-CD3 antibody (IC50 = 2267 nM) than it is in inhibiting human B cell proliferation mediated by BCR activation (IC50 = 2 nM). The compound appears to be more potent in its ability to block anti-CD28–induced human T cell proliferation, but its potency in this assay (IC50 = 124 nM) is still approximately 60 times less than that in the BCR-induced B cell proliferation assay. When the proliferation of human T cells was stimulated by TCR activation through both CD3 and CD28, an activation paradigm most relevant to the in vivo setting, the potency of INCB040093 (IC50 = 1866 nM) was again similar to that seen when the TCR was activated with anti-CD3 alone. Furthermore, INCB040093 was even less potent in inhibiting IL-2–induced T cell proliferation, with an IC50 value of 3107 nM.
Compared with inhibition of B cell proliferation, INCB040093 is also less potent in inhibiting IL-2–induced NK cell proliferation, fMLP-induced elastase exocytosis in neutrophils, and LPS-induced TNF-α production in PBMCs, with IC50 values of 708, 1566, and 2704 nM, respectively (Table 1). In addition, INCB040093 was inactive in blocking CC chemokine receptor (CCR)2-mediated chemotaxis. Taken together, these results suggest that the impact of INCB040093 on the human immune system will likely be preferential to B cells and Th17 cells.
INCB040093 Is Active in Inhibiting Proliferation of and PI3K Signaling in Cell Lines Derived from B Lymphoid Malignancies.
Given its potent inhibitory activity against BCR-induced human B cell proliferation, INCB040093 was tested for its effects on proliferation, as measured by [3H]thymidine uptake, of a number of cell lines derived from human B cell malignancies. As shown in Table 2, several cell lines from three different B cell malignancy subtypes [diffuse large cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and HL] showed meaningful sensitivity to INCB040093 (IC50 < 100 nM). No inhibitory effects were observed on a panel of 24 cell lines of non-B cell origin at a 10 μM concentration (data not shown). These results suggested that INCB040093 could have preferential therapeutic activity in B cell malignancies in humans. For comparison, idelalisib was evaluated in the same study. It showed similar activity as INCB040093 in inhibiting proliferation of the DLBCL cell lines, but was more potent against the cell lines of MCL (Table 2).
Inhibitory activity of INCB040093 against proliferation of cell lines derived from B cell malignancies
Akt phosphorylation is a major signaling event downstream of PI3K activation. In addition, it is well established that activated Akt can phosphorylate FOXO transcriptional factors, which prevent these cell cycle inhibitors from moving into the nucleus, thereby limiting their growth-inhibitory functions (Fabre et al., 2005). To test the hypothesis that INCB040093 could inhibit the proliferation of several B cell lines by targeted inhibition of PI3Kδ signaling, the effects of INCB040093 on pAkt and pFOXO levels in Pfeiffer cells (DLBCL) were evaluated. The Pfeiffer cell line was chosen because it is one of the most sensitive cell lines to INCB040093 for growth inhibition and contains detectable levels of PI3Kδ as well as constitutive pAkt and pFOXO proteins. As shown in Fig. 3, INCB040093 inhibits both pAkt and pFOXO levels in the Pfeiffer cells in a dose-dependent fashion, as detected by Western blot analysis, with IC50 values of approximately 10 nM, consistent with its potency in blocking cell proliferation. Because INCB040093 has similar potency in inhibiting proliferation and PI3Kδ signaling in the Pfeiffer cells, it suggests that the proliferation of this particular cell type is driven largely by constitutive signaling of PI3Kδ and provides a mechanism by which INCB040093 may inhibit the proliferation of Pfeiffer and other B cell lines.

INCB040093 potently inhibits constitutive PI3K signaling in Pfeiffer cells of DLBCL. The Pfeiffer cells were treated with different amount of INCB040093 for 2 hours. The levels of pAkt (Ser473) and pFOXO3a (T32)/pFOXO1 (T24) in the total cell lysates were then measured by Western blotting (A and B). The Pfeiffer cells were also spiked into human whole blood and treated with INCB040093. The level of pAkt (Ser473) in the Pfeiffer cells was evaluated by intracellular staining with an anti-pAkt (Ser473) antibody, followed by fluorescence-activated cell sorting analysis (C).
The functional effectiveness of INCB040093 in blocking PI3Kδ signaling in malignant B cells was further evaluated in human whole blood. Pfeiffer cells were added to human whole blood and incubated with a range of INCB040093 concentrations for 1 hour. Levels of pAkt in the Pfeiffer cells were then measured by flow cytometry using the intracellular staining intensity of an Alexa-488–conjugated anti-pAkt antibody as the readout. As shown in Fig. 3C, INCB040093 inhibited pAkt level in the Pfeiffer cells with an IC50 value of 71 nM. This is consistent with the plasma protein binding (fraction unbound in human plasma = 18.6%)-adjusted potency of the compound in the cell proliferation and signaling assays (protein binding-adjusted IC50 = 53 nM; protein binding-adjusted IC90 ∼400 nM).
INCB040093 Induces Apoptosis and Reduces Viability in Cell Lines of B Cell Malignancy.
In addition to its inhibitory effect on normal and malignant B cell proliferation, INCB040093 was evaluated for its ability to induce cell apoptosis in B cell lines. As shown in Fig. 4, INCB040093 treatment at 1 μM for 24 hours resulted in approximately twofold increase in annexin V staining in the SU-DHL-5, Pfeiffer, and WSU-NHL cell lines, indicating a significant level of apoptosis induction in these cells. The 1 μM dose level was chosen based on the previous finding from the signaling study that INCB040093 at this dose level did not affect signaling function of PI3Kα or PI3Kβ.

INCB040093 induces apoptosis in DLBCL cell lines. SU-DHL-5, Pfeiffer cells, and WSU-NHL cells were treated with or without INCB040093 at 1 μM for 24 hours. The percentage of apoptotic cells was determined by annexin V-fluorescein isothiocyanate and propidium iodide staining, followed by two-color flow cytometric analysis. Percentages represent both annexin V single-positive and annexin V/propidium iodide double-positive cells.
INCB040093 was also tested for its effect on the viability of the Pfeiffer cell line. As shown in Fig. 5, A and B, INCB040093 by itself could dose-dependently reduce the viability of the Pfeiffer cells. Interestingly, it was observed that the inhibitory effect of INCB040093 on the viability of the Pfeiffer cells was much reduced by the presence of IL-10 in the culture medium (Fig. 5, A and B). Concurrently, it was shown that IL-10 could signal into the Pfeiffer cells through phosphorylation of STAT3 (Fig. 5C). Because JAK is the upstream kinase that directly phosphorylates and activates STAT3, it was hypothesized that JAK inhibition could relieve the inhibitory effect of IL-10 on the ability of INCB040093 to reduce the viability of the Pfeiffer cells. To test this hypothesis, two JAK inhibitors were used. Ruxolitinib (also known as INCB018424) is a JAK1/2 inhibitor with IC50 values against JAK1 and JAK2 of 3.3 nM and 2.2 nM (Quintás-Cardama et al., 2010). Itacitinib (also known as INCB039110) is a JAK1 selective inhibitor with IC50 against JAK1 of 2.3 nM (Incyte internal observation). Indeed, as shown in Fig. 5, A and B, either JAK1/2 inhibition with ruxolitinib or JAK1 inhibition with itacitinib reversed the suppressive effect of IL-10 on the function of INCB040093 in reducing the viability of the Pfeiffer cells.

IL-10–induced Pfeiffer cell resistance to INCB040093 in viability assay can be overcome by inhibition of JAK. The Pfeiffer cells were treated with different amount of INCB040093 for 3 days in the absence or presence of IL-10 (10 ng/ml), INCB018424 (Ruxolitinib) (1 μM, JAK1/2 inhibitor) (A), or INCB039110 (itacitinib) (1 μM, JAK1 selective inhibitor) (B). The cell viability was assessed using a cell titer-grow assay. Phosphorylation of STAT3 induced by IL-10 (10 ng/ml), IL-6 (10 ng/ml), HS-5 stroma cell conditioned medium (SCM) in the Pfeiffer, and other DLBCL cell lines was evaluated by Western blotting (C).
INCB040093 May Change the Tumor Microenvironment by Inhibiting Tumor Cell Production of Cytokines and Chemokines That Are Important for Stroma Cell Function.
In clinical studies, a relatively high response rate to INCB040093 treatment was observed in patients with HL (Phillips et al., 2015). In agreement, as shown in Table 3, proliferation of HS-445, a HL cell line, was found to be sensitive to INCB040093 (IC50 = 75 nM). However, Reed-Sternberg (RS) cells (the malignant B cell in HL tumor mass) often represent less than 2% of the tumor mass. Inflammatory cells comprised of a large portion of the HL tumor and the tumor microenvironment appear to be essential for RS cell survival and growth in HL (Aldinucci et al., 2010). A previous study showed that stroma cells could promote PI3Kδ signaling in HL cells. Conversely, PI3Kδ activity in HL cells could enhance production of CCL5, a stroma-stimulating chemokine (Meadows et al., 2012). To examine the potential impact of PI3Kδ inhibition by INCB040093 on HL cells in the tumor microenvironment, HS-445 cells were cocultured for 3 days in different ratio with HS-5 cells, a human bone marrow stromal cell line, in the presence or absence of INCB040093 (1 μM). The supernatants were collected, and levels of a broad panel of cytokines and chemokine were measured. As shown in Fig. 6, the compound treatment reduced production of TNF-β and (MIP)-1β, a CCL5-related chemokine from the HS-445 cells. Both cytokines have been shown to either directly or indirectly impact immune function.
Pharmacokinetics of INCB040093 in rats following a 5 mg/kg i.v. and a 10 mg/kg PO

INCB040093 reduces TNF-β (A) and MIP-1β (B) production in a coculture of Hodgkin’s lymphoma cell line (HS-445) and a stroma cell line (HS-5). HS-445 and HS-5 cells were cocultured for 3 days in different ratios in the presence or absence of INCB040093 (1 μM). The levels of cytokines and chemokines in the supernatants were evaluated using a Luminex assay specific for a broad panel of human cytokines and chemokines.
INCB040093 Suppresses Tumor Growth in Pfeiffer Model of Lymphoma.
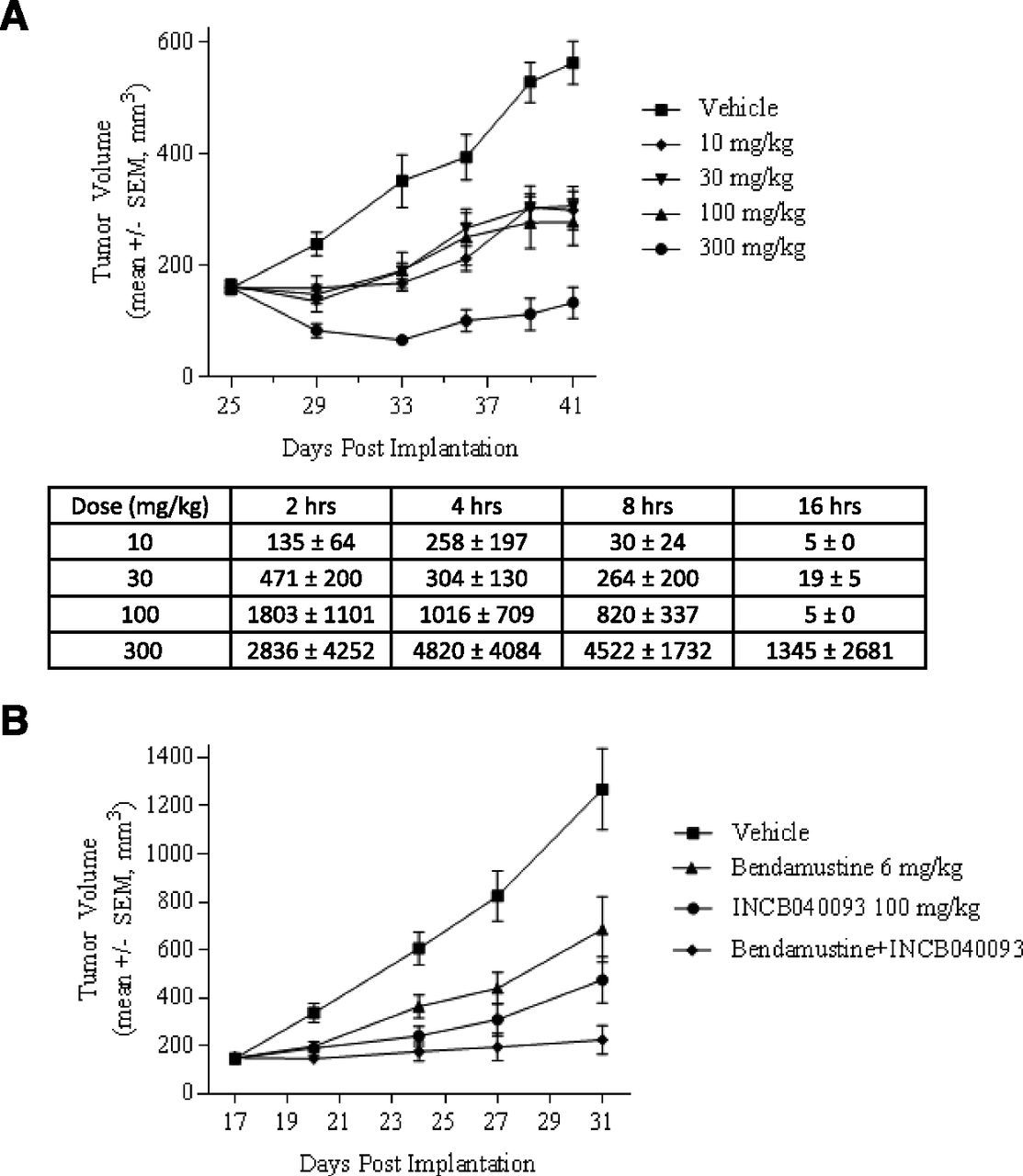
Pfeiffer cells are exquisitely sensitive to INCB040093 in vitro, and we hypothesized that Pfeiffer xenografts would be a robust model for testing the effects of this compound in vivo. INCB040093 was evaluated as a single agent or in combination with bendamustine (Treanda), a cytotoxic DNA damaging agent approved for use in lymphoid malignancies. SCID mice bearing well-established tumors were treated with twice-daily PO of vehicle or INCB040093 at 10, 30, 100, and 300 mg/kg for 16 days, and tumor volumes were measured twice weekly. INCB040093 treatment resulted in statistically significant tumor growth inhibition of 44%, 47%, 50%, and 80% (P < 0.001) at doses of 10, 30, 100, and 300 mg/kg, respectively (Fig. 7A).

INCB040093 is effective in suppressing tumor growth in the Pfeiffer mouse model of DLBCL. The Pfeiffer cells were s.c. inoculated into the immune-compromised SCID mice. After 2–3 weeks, these cells formed tumors with volumes exceeding 100 mm3. The mice with inoculated Pfeiffer cells were randomized into treatment groups to receive twice-daily PO of vehicle or INCB040093 at 10, 30, 100, and 300 mg/kg for 16 days. Mean plasma concentration (nanomolar) of INCB040093 was measured at various times following repeated twice-daily PO in the Pfeiffer tumor xenograft model. (A) Mice bearing s.c. Pfeiffer tumors were also randomized into treatment groups that received vehicle, INCB040093 (100 mg/kg, oral twice daily), bendamustine (6 mg/kg, i.p., 4 consecutive days each week), or the combination of INCB040093 and bendamustine (B). Tumor volumes were ascertained approximately twice each week.
Steady state plasma samples were collected from these mice to determine the circulating levels of INCB040093. In these studies, the 16-hour time point is considered to be the trough of the dosing interval. The targeted clinical trough concentration was ∼400 nM, which is the human serum protein binding-adjusted IC90 for the Pfeiffer cell proliferation. Based on the data in Fig. 7A, optimal tumor growth inhibition was observed at a dose that exceeded the proposed clinical plasma trough concentration of INCB040093 throughout the day. However, efficacy was also observed at dose levels that exceeded the human serum protein binding-adjusted IC50 (∼70 nM) for greater than 4 hours each day, although a clear dose response was not observed.
The effect of INCB040093 on tumor growth in combination with relevant cytotoxic chemotherapy was also assessed. Mice bearing s.c. Pfeiffer tumors were randomized into treatment groups that received vehicle, INCB040093 (100 mg/kg, oral twice daily), bendamustine (6 mg/kg, i.p., 4 consecutive days each week), or the combination. The dose levels of INCB040093 and bendamustine were each below the maximally effective doses, a common practice when exploring the potential for additive or synergistic effects of two agents in preclinical models. Tumor growth inhibition at the end of the 2-week dosing period was 46% for bendamustine, 62% for INCB040093, and 82% for the combination of both compounds (Fig. 7B). No significant (>10%) body weight loss was noted in any treatment group. These data suggest that selective inhibition of PI3Kδ can potentiate the antitumor effects of bendamustine, a clinically relevant chemotherapeutic, in vivo without obvious toxicity.
INCB040093 Is a Suitable Candidate for Clinical Development.
The pharmacokinetic parameters for INCB040093 in male Sprague–Dawley rats were measured, and the results are summarized in Table 3. The mean systemic plasma clearance of INCB040093 determined following the i.v. dose was moderate (1.42 l/h per kilogram), approximately 43% of the hepatic blood flow (3.3 l/h per kilogram). The mean volume of distribution was 1.52 l/kg, indicating that INCB040093 is distributed beyond the total body volume of body water in rats (0.7 l/kg). The mean terminal half-life was 5.3 hours. The average renal clearance of INC040093 (0.00605 l/h per kilogram) was much less than the glomerular filtration rate (0.31 l/h per kilogram). Less than 1% of the administered i.v. dose was excreted in urine as parent compound.
Following oral administration, the mean Cmax and Tmax were 4.82 μM and 0.5 hour, respectively, and the mean area under the curve was 8.84 μM × h. The apparent mean terminal half-life after oral dosing was 1.9 hours. The mean oral bioavailability of INCB040093 in rats was 53.5%.
Furthermore, INCB040093 was evaluated for potential toxicity in rats and Beagle dogs following administration for up to 3 months. Reversible hematologic effects and decreased cellularity or loss of germinal centers in lymphoid tissues, an expected pharmacologic effect resulting from PI3Kδ inhibition, were observed in rats and dogs. Generalized depletion of bone marrow occurred at low incidence only at high doses. In rats, hair loss and scabbing were observed only in the 3-month study, which was considered likely secondary to the immunologic effects of INCB040093. Liver toxicity, including elevated alanine aminotransferase, aspartate aminotransferase, sorbitol dehydrogenase, alkaline phosphatase, and gamma glutamyl transferase, and microscopic findings of inflammation and hepatocellular necrosis, was observed in dogs. Exposures at the NOAEL (no-observed-adverse-effect-level) for liver findings in dogs exceeded plasma levels associated with clinical exposures, indicating an acceptable safety margin. INCB040093 was not genotoxic in a standard battery of in vivo and in vitro studies.
Discussion
In this work, we have disclosed the chemical structure of INCB040093 and demonstrated that INCB040093, which is currently under consideration for further clinical studies for treatment of B cell malignancies, is a novel, potent, and selective inhibitor of the PI3Kδ isoform. Its activity against and selectivity for the class 1 PI3K isoforms were first assessed with biochemical enzyme assays and then further evaluated with the PI3K isoform-specific signaling assays in cells. The cellular activity of INCB040093 against PI3Kδ functions was extensively investigated by utilizing assays that measure the biologic consequences of PI3Kδ inhibition in primary leukocytes from different species and in cell lines of B cell malignancies. The detailed pharmacokinetic and safety profiles of INCB040093 in rats and dogs and its pharmacological efficacy in the Pfeiffer model for DLBCL are also described.
We have shown that INCB040093 is a very potent inhibitor of the proliferation of primary human and dog B cells induced by PI3Kδ activation downstream of the B cell receptor, a Toll-like receptor, cytokine receptors, and a costimulatory receptor. INCB040093 is also potent in suppressing proliferation of cell lines of various B malignancies in vitro and tumor growth in the Pfeiffer model for DLBCL in vivo. The mechanism by which INCB040093 inhibits proliferation of cell lines of B cell malignancies was addressed by investigating its effect on PI3K signaling in these cells. As shown in this study, the compound potently inhibits constitutive PI3K signaling, as measured by pAkt and pFOXO in the Pfeiffer cells.
There is now strong evidence for a potential causative role of excessive PI3K activity in the development of B cell malignancies. For example, mice expressing a constitutively active form of PI3K develop infiltrating lymphoproliferative disorders (Borlado et al., 2000). Activation of PI3K pathway was demonstrated in DLBCL cell lines and primary cells in DLBCL patients (Uddin et al., 2006), MCL cell lines and primary cells in MCL patients (Rudelius et al., 2006), follicular lymphoma (Garcia-Martinez et al., 2011), and mediastinal large B cell lymphoma (Renne et al., 2007). Within the class I PI3K isoform family, several studies have found enhanced PI3Kδ activity and therefore suggest a role of PI3Kδ signaling in the pathogenesis of different B cell malignancies, including CLL (Herman et al., 2010), HL (Meadows et al., 2012), and plasma cell myeloma (Ikeda et al., 2010). These findings in the literature regarding PI3K signaling in malignant B cells and the data presented in this report suggest that INCB040093 inhibits the proliferation of B cell lines by reducing constitutive PI3K signaling in these cells.
However, given the complexity in pathogenesis of B cell malignancies, it will be too simplistic a view to expect all the malignant B cells to have the same level of dependency on constitutive PI3K signaling for cell proliferation. In this study we found that only about half of the DLBCL cell lines tested was sensitive to INCB040093 inhibition in the proliferation assay. We do not know the mechanism(s) behind the resistance to INCB040093 in these DLBCL cell lines at this time. We believe that comprehensive profiling of genetic makeups of the sensitive versus resistant tumors in the current clinical studies would help address this question and provide guidance for future patient selection.
We also show that INCB040093 can induce apoptosis in three different DLBCL cell lines and reduce viability of the DLBCL cell line, Pfeiffer. Interestingly, the ability of INCB040093 to reduce the viability of the Pfeiffer cells is suppressed by the presence of IL-10 in the culture medium. IL-10 is present in the serum of patients with DLBCL, and its level has been shown to be a prognostic factor for these patients. IL-10 is known to signal through the JAK/STAT pathway, which is confirmed by this study showing IL-10 induces phosphorylation of STAT3 in multiple DLBCL cell lines. In this study we show that either JAK1/2 or JAK1 inhibition can reverse the suppressive activity of the cytokine on the ability of INCB040093 to reduce the viability of the Pfeiffer cells, suggesting combined inhibition of PI3Kδ and JAK1 would have a better effect in treating DLBCL than PI3Kδ inhibition alone. Consistent with this hypothesis, better clinical response was observed in patients with HL when treated with INCB040093 plus the JAK1 inhibitor itacitnib than INCB040093 alone (Phillips et al., 2015).
The tumor microenvironment can have profound effects on tumor cell growth and survival. A previous study using cocultures of CLL cells and nurse-like stroma cells suggests that PI3Kδ inhibition cannot only antagonize intrinsic survival signals in tumor cells but also block survival signals provided by extrinsic factors in the tumor microenvironment (Hoellenriegel et al., 2011). We found that INCB040093 can have a strong inhibitory effect on the differentiation of Th17 cells with IC50 values less than 10 nM. Th17 cells are inflammatory and could mediate tumor growth in two distinct ways—by driving angiogenesis and by suppressing antitumor immunity (Bailey et al., 2014). It remains to be determined whether the anti-Th17 effect of INCB040093 could be part of the mechanism for its anti-B cell malignancy effect in clinical studies. In addition, INCB040093 can also reduce MIP-1β and TNF-β production from HS-445, a HL cell line. This result is pertinent to the observed clinical efficacy of INCB040093 in HL patients and similar to a previous report in which PI3Kδ inhibition by idelalisib inhibited HL cell production of CCL5 (Meadows et al., 2012), which is related to MIP-β as both chemokines are ligands for CCR5. HL is unique among all cancers because malignant RS cells are greatly outnumbered by stroma cells in tumor microenvironment. RS cells in HL are known to secrete different chemokines and cytokines, including CCR5 ligands and TNF-β, to help nurture stroma cells so that the cancer cells can proliferate, survive, and evade host immune attack (Steidl et al., 2011). It is conceivable that INCB040093 is effective for treating HL by not only blocking HL cell proliferation directly but also by inhibiting CCR5-mediated chemotaxis of stroma cells into the tumor mass and TNF-β–dependent survival of stroma cells.
PI3Kδ has been implicated in the development and/or function of T cells, NK cells, and neutrophils. In mouse T cells, PI3Kδ was suggested to be the main PI3K isoform that generates PIP3 downstream of the T cell receptor (TCR) and CD28, a costimulatory receptor, although PI3Kα, PI3Kβ, and PI3Kγ are also likely to contribute (Vanhaesebroeck et al., 1999; Okkenhaug et al., 2002, 2006). Mice with an inactive PI3Kδ kinase were found to have reduced total NK cellularity, although the cytotoxicity of NK cells was only marginally impaired (Guo et al., 2008). Although evidence for the involvement for PI3Kδ in mouse neutrophil function has been controversial due to differences in phenotypic responses when studied in PI3Kδ kinase knockout mice, in kinase knock-in mice, and using small-molecule inhibitors of PI3Kδ (Rommel et al., 2007), one report did demonstrate that inhibition of PI3Kδ could reduce TNF-α– and fMLP-induced superoxide generation and elastase exocytosis in human neutrophils (Sadhu et al., 2003). To evaluate potential immune toxicity that can result from INCB040093 treatment of B cell malignancies, INCB040093 was tested for its ability to inhibit proliferation of human T cells induced by anti-CD3 and anti-CD28 antibodies. The effects of INCB040093 on IL-2–induced proliferation of human NK cell and T cells, fMLP-induced elastase exocytosis in human neutrophils, and LPS-induced TNF-α production in human PBMCs were also evaluated. In addition, because CCR2-mediated chemotaxis has been reported to be dependent on PI3Kγ-mediated signaling (Sasaki et al., 2000), the effect of INCB040093 on this function in human monocytes was examined as a selectivity control. The inhibitory activities of INCB040093 against the proliferation or function of T cells, NK cells, neutrophils, and monocytes measured in this study were much weaker than those against B cells, with the exception of Th17 cell differentiation. These results suggest that INCB040093 is potentially useful in reducing tumor growth of B cell malignancies without profound suppressive effect on other immune cell types.
As exemplified by the pharmacokinetic study in rats, NCB040093 has a favorable drug-like pharmacokinetic profile. The results from the 3-month safety studies in dogs and rats are also in favor of advancement of INCB040093 into clinical studies. Idelalisib, the only PI3Kδ inhibitor in the market, is approved for the treatment of patients with relapsed chronic lymphocytic leukemia and follicular lymphoma. INCB040093 has demonstrated a good clinical efficacy in treating patients with HL in phase 1 studies. INCB040093 also has demonstrated antitumor effect, either as monotherapy or in combination with bendamustine, in the Pfeiffer model of DLBCL, suggesting that the compound may be useful for treating patients with DLBCL. Thus, INCB040093 could have different utility than idelalisib.
In summary, data collected in the in vitro and in vivo preclinical studies described in this work clearly support further clinical development of INCB040093 for treating different types of B cell malignancies.
Authorship Contributions
Participated in research design: Shin, Y.-L. Li, Soloviev, Diamond, Newton, Scherle.
Conducted experiments: K. H. Wang, Hall, Katiyar, Q. Wang, Yang, Rumberger, Leffet, He, Rupar, Bowman, Favata, J. Li, Covington, Shin.
Contributed new reagents or analytic tools: Y.-L. Li, Mei.
Performed data analysis: Shin, Covington, Liu, Y. Li, Koblish, Soloviev, Shuey, Combs, Yao, Diamond, Fridman, Burn, Yeleswaram, Hollis, Vaddi, Huber, Newton, Scherle.
Wrote or contributed to the writing of the manuscript: Shin, Koblish, Shuey, Covington, Scherle.
Footnotes
- Received September 1, 2017.
- Accepted October 31, 2017.
All authors are current or former employees of Incyte and may hold equity in Incyte.
This work was supported entirely by Incyte.
Abbreviations
- BAFF
- B cell activating factor
- BCR
- B cell antigen receptor
- CCL
- CC chemokine ligand
- CCR
- CC chemokine receptor
- DLBCL
- diffuse large cell lymphoma
- DMAC
- dimethylacetamide
- FBS
- fetal bovine serum
- fMLP
- N-formyl peptide
- HL
- Hodgkin’s lymphoma
- IL
- interleukin
- JAK
- Janus kinase
- LPA
- lysophosphatidic acid
- LPS
- lipopolysaccharide
- MCL
- mantle cell lymphoma
- MIP
- macrophage-inflammatory protein
- NK
- natural killer
- PBMC
- peripheral blood mononuclear cell
- PBS
- phosphate-buffered saline
- PDGF
- platelet-derived growth factor
- PI3K
- phosphatidylinositol 3-kinase
- PIP3
- phosphatidylinositol-3,4,5-triphosphate
- PO
- oral dose
- RBC
- red blood cell
- RS
- Reed-Sternberg
- STAT
- signal transducer and activator of transcription
- TCR
- T cell antigen receptor
- Th
- T helper
- TNF
- tumor necrosis factor
- Copyright © 2017 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}