


Abstract
Therapies targeting either interleukin (IL)-23 or IL-17 have shown promise in treating T helper 17 (Th17)–driven autoimmune diseases. Although IL-23 is a critical driver of IL-17, recognition of nonredundant and independent functions of IL-23 and IL-17 has prompted the notion that dual inhibition of both IL-23 and IL-17 could offer even greater efficacy for treating autoimmune diseases relative to targeting either cytokine alone. To test this hypothesis, we generated selective inhibitors of IL-23 and IL-17 and tested the effect of either treatment alone compared with their combination in vitro and in vivo. In vitro, using a novel culture system of murine Th17 cells and NIH/3T3 fibroblasts, we showed that inhibition of both IL-23 and IL-17 completely suppressed IL-23–dependent IL-22 production from Th17 cells and cooperatively blocked IL-17–dependent IL-6 secretion from the NIH/3T3 cells to levels below either inhibitor alone. In vivo, in the imiquimod induced skin inflammation model, and in the myelin oligodendrocyte glycoprotein peptide–induced experimental autoimmune encephalomyelitis model, we demonstrated that dual inhibition of IL-17 and IL-23 was more efficacious in reducing disease than targeting either cytokine alone. Together, these data support the hypothesis that neutralization of both IL-23 and IL-17 may provide enhanced benefit against Th17 mediated autoimmunity and provide a basis for a therapeutic strategy aimed at dual targeting IL-23 and IL-17.
Introduction
The IL-23/IL-17–mediated axis of inflammation is pivotal to the development of T cell–driven autoimmune diseases, including rheumatoid arthritis, inflammatory bowel disease, ankylosing spondylitis, multiple sclerosis (MS), psoriasis, and psoriatic arthritis (Chen et al., 2006; Uyttenhove and Van Snick, 2006; Gaffen et al., 2014). IL-23/IL-17–mediated pathology is associated with a subset of CD4+ effector T cells called T helper 17 (Th17) (Harrington et al., 2006). The pathogenic function of Th17 cells is driven in part by IL-23, a heterodimeric cytokine consisting of a p19 subunit coupled to the p40 subunit that is also shared by IL-12. IL-23 promotes Th17 pathogenesis by driving expression of the hallmark Th17 cytokine, IL-17, as well as other cytokines, such as IL-22 and granulocyte monocyte colony-stimulating factor (GM-CSF), and by suppressing IL-10 (Murphy et al., 2003; Langrish et al., 2005; McKenzie et al., 2006; McGeachy et al., 2007; McGeachy, 2011). IL-17 (or IL-17A) is the founding member of a family of six structurally related cytokines named IL-17A through F (Aggarwal and Gurney, 2002; Weaver et al., 2007). The most closely related IL-17 members, IL-17A and IL-17F, can be secreted as IL-17A/A and IL-17F/F homodimers as well as an IL-17A/F heterodimer (Chang and Dong, 2007). All three are produced by Th17 cells but, arguably, IL-17A/A and IL-17A/F are the more proinflammatory members (Ishigame et al., 2009). In addition to Th17 cells, IL-17 can be produced by CD8+ T cells, γδ T cells, and invariant natural killer T (iNKT) cells and by lymphoid tissue inducer cells (Cua and Tato, 2010). IL-17 triggers fibroblasts, epithelial cells, endothelial cells, and macrophages to secrete cytokines, chemokines, and growth factors, including IL-6, IL-8, CXCL1, granulocyte colony-stimulating factor, and GM-CSF, which recruit and expand neutrophils as well as monocytes and enhance the local inflammatory milieu (Aggarwal and Gurney, 2002). Acknowledgment of the critical involvement of IL-23 and IL-17 in autoimmune disorders has motivated the discovery of therapeutics for targeting IL-23 and IL-17 pathways.
Many pharmaceutical companies have now developed antibodies against IL-23 or IL-17 that have been largely successful in clinical trials, but they have also presented some unexpected results as well. Antibodies against the common p40 subunit shared by IL-12 and IL-23 and IL-23–specific antibodies were remarkably efficacious in psoriasis patients and showed promising results in Crohn’s disease. However, in MS patients, the trials with anti-p40 antibodies failed (Segal et al., 2008; Longbrake and Racke, 2009; Sandborn et al., 2012; Kimball et al., 2013; Papp et al., 2013a; Gaffen et al., 2014). Agents that block IL-17 were impressively efficacious in psoriasis, psoriatic arthritis, and ankylosing spondylitis, and elicited disease improvement in rheumatoid arthritis patients (Leonardi et al., 2012; Papp et al., 2012a,b, 2013b; Kellner and Kellner, 2013; Patel et al., 2013; Rich et al., 2013; Sigurgeirsson et al., 2014). However, trials with IL-17 inhibitors in Crohn’s disease were prematurely terminated owing to a lack of efficacy and in some cases an exacerbation of the disease (Hueber et al., 2012). These mixed outcomes highlight the complexity of the IL-23–IL-17 pathways and underscore the fact that IL-23 and IL-17 are not entirely redundant targets.
Although IL-23 drives IL-17 production from Th17 cells, IL-23 and IL-17 can function independently of each other. IL-23 induces additional proinflammatory cytokines such as IL-22 and GM-CSF, which have been shown to be more critical than IL-17 to the pathology of certain animal disease models (Liang et al., 2006; Zheng et al., 2007; van der Fits et al., 2009; Codarri et al., 2011; El-Behi et al., 2011). IL-17 can be induced independently of IL-23 by transforming growth factor (TGF)-β and IL-6 in mice (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006) and by T-cell receptor activation in human cells (unpublished observation). Other cell types can also be stimulated to produce IL-17 without IL-23, such as iNKT cells by α-galactosylceramide, and mast cells with complement component 5a, lipopolysaccharide, tumor necrosis factor-α or IgG complexes (Yoshiga et al., 2008; Hueber et al., 2010).
Neutralization of IL-17 alone might block all IL-17 regardless of its source, but other IL-23–dependent cytokines such as IL-22 and GM-CSF would be unaltered and could contribute to disease. Blockade of IL-23 alone would be expected to inhibit all IL-23–dependent cytokines including much of the Th17-produced IL-17. However, not all pathogenic IL-17 would be inhibited since it can be produced without IL-23 in certain situations. Therefore, dual inhibition of IL-23 and IL-17 could neutralize the IL-23–dependent cytokines as well as the effects of IL-23–independent IL-17 leading to a comprehensive blockade of the IL-23/IL-17 pathway and enhanced efficacy in treating disease.
Targeting a more complete neutralization of the IL-23/IL-17 pathway has therefore been of interest. Although agents targeting both IL-17 and IL-23 have been reported, a complete understanding of the potential for improved efficacy with dual inhibition of IL-23 and IL-17 is lacking (Mabry et al., 2010). To investigate if greater efficacy could be achieved with dual blockade, we generated potent and selective inhibitors of IL-23 and IL-17A. These tools enabled us to explore the impact of IL-23 and IL-17 inhibition alone and in combination in vitro and in vivo. We show in a novel in vitro culture system, where the impact of IL-23 and IL-17 blockade could be tested within the same setting, that dual inhibition caused a stronger suppression of downstream cytokines than blockade of either cytokine alone. Furthermore, combination treatment with both IL-23 and IL-17 inhibitors in vivo offered superior efficacy compared with treatment with either inhibitor alone in the imiquimod (IMQ)-induced skin inflammation and the myelin oligodendrocyte glycoprotein (MOG) peptide–induced experimental autoimmune encephalomyelitis (EAE) mouse models.
Materials and Methods
Mice.
BALB/c mice were purchased from either Charles River Laboratories (Wilmington, MA) or Harlan Laboratories (Indianapolis, IN) and C57BL/6 mice were purchased from Charles River, Harlan Laboratories, or The Jackson Laboratories (Bar Harbor, ME). The strain, sex, age and vendor of the mice used for each in vivo protocol is indicated within the description of each procedure below. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee.
Generation of IL-23p19 Mouse Adnectin.
The ATI-1221 anti-mouse IL-23p19 adnectin was generated using established methods (Mitchell et al., 2014). Briefly, murine IL-23 [eBioscience, San Diego, CA; chemically biotinylated at Bristol-Myers Squibb (BMS; Princeton, NJ)] was presented to large synthetic libraries of adnectins using murine anti-p40 mAbs (clone C17.8; eBioscience). In this way, the IL-23–specific p19 subunit was displayed for adnectin binding. To generate the pegylated version of the adnectin, ATI-1249, purified ATI-1221, was conjugated with a maleimide derivative of a 40-kDa 2-branched polyethylene glycol moiety (mPEG2-MAL) via standard maleimide chemistry at the penultimate C-terminal cysteine residue according to methods described previously (Mitchell et al., 2014).
Generation of Mouse IL-17RA-Fc Decoy Receptor.
Mouse IL17RA-Fc (mIL-17RA-Fc) was overexpressed in transiently transfected HEK293-6E cells. Conditioned medium was loaded to protein A column. Captured protein was eluted by 50 mM sodium acetate buffer, pH 3.5. The eluate was neutralized by 1M Tris immediately. Protein was further purified by Superdex 200 size-exclusion column to remove aggregates, and exchange buffer to phosphate buffered saline (PBS), pH 7.2.
Generation of Mouse IL-17 Monoclonal Antibody.
BALB/c mice (Charles River Laboratories) were immunized intraperitoneally and subcutaneously four times with recombinant murine (rm) IL-17A (R&D Systems/Bio-Techne, Minneapolis, MN) (2 μg/mouse per injection) in Ribi adjuvant (Sigma-Aldrich, St. Louis, MO). Serum samples were evaluated for reactivity to the immunogen by enzyme-linked immunosorbent assay (ELISA) using rmIL-17 (R&D Systems). The mice received two final boosts of 2 μg/mouse per injection of rmIL17 on days 3 and 2 before fusion. B cells from spleens harvested from mouse #230891 were then fused (ratio of 1:1) with mouse ×63.Ag8.653 myeloma cells (CRL-1581; American Type Culture Collection, Manassas, VA) by electrofusion (Hybrimmune, ECM 2001; Harvard Apparatus, Holliston, MA). The resulting hybridomas were plated out in flat-bottom 96-well plates (200 μl/well), and incubated for 7 days in hypoxanthine-aminopterin-thymidine (HAT) selection medium [Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 5% BM-Condimed H1 Hybridoma Cloning Supplement (Roche, Atlanta, GA), and 1× HAT] and an additional 7 days in 1× sodium hypoxanthine and thymidine selection medium (same medium supplemented with 1× sodium hypoxanthine and thymidine instead of HAT). Hybridoma supernatants were then screened by ELISA for antigen specificity (see below). Antigen-specific clones were subcloned by limiting dilution to achieve stability and monoclonality. Clones of interest were expanded and antibodies were purified from culture supernatant over protein A column. Hybridoma clones were preserved by freezing in 10% DMSO freezing media (Sigma-Aldrich).
ELISA Screening for Antigen-Specific Detection of Anti–mIL-17A mAbs.
Recombinant mouse IL-17 (R&D Systems) was coated on a 96-well plate at 2 μg/ml overnight at 4°C (50 μl/well). The plate was blocked with 1× PBS solution with Tween (PBST) containing 1% bovine serum albumin (200 μl/well) for 1 hour at room temperature, then washed three times with 1× PBST. Undiluted hybridoma supernatants were added to each well (50 μl/well). Plates were washed three times with 1× PBST before addition of anti-mouse IgG-Fc–specific horseradish peroxidase–conjugated antibody (Jackson ImmunoResearch, West Grove, PA) diluted 1:2000 in 1× PBST with 1% bovine serum albumin (50 μl/well). Plates were incubated with the secondary antibody for 1 hour at room temperature. The plates were washed six times prior to addition of 100 μl/well of ABTS substrate (Moss Inc., Pasadena, MD), and absorbance was read at 450 nm and referenced at 620 nm. Rat anti-mouse IL-17 antibodies (clone 50104 and clone 50101; R&D Systems) were used as positive control in the ELISA assay along with irrelevant rat IgG (Sigma-Aldrich) and mouse IgG (Sigma-Aldrich) as isotype controls.
Mouse IL-23–Dependent Phospho-STAT3 Induction in Kit225 Cells.
Kit225 cells were seeded into 96-well plates at 4 × 105 cells/well and were rendered quiescent in the absence of fetal bovine serum and IL-2 for 3 hours at 37°C. For inhibition dose responses for ATI-1221 and ATI-1249, 10 pM rmIL-23 (generated at BMS), or 10 pM rmIL-23 preincubated with a range of concentrations of ATI-1221 and ATI-1249 antagonist for 1 hour, was applied to the cells and the cells returned to the incubator for 15 minutes at 37°C to allow the phosphorylation of STAT3 (abbreviated as pSTAT3). Each test condition was assayed in duplicate in 96-well plates. Stimulation was stopped by placing the cells on ice and adding ice-cold PBS. Finally, the cells were pelleted and lysed following standard protocols and pSTAT3 production detected by ELISA (Cell Signaling Technology, Danvers, MA). Results expressed as percent inhibition of pSTAT3 levels and IC50s were determined using GraphPad Prism software (GraphPad Software, San Diego, CA).
Mouse IL-23–Dependent Cytokine Induction in Primary Th17 Cells.
To differentiate mouse Th17 cells for analysis, CD4+ T cells were enriched from the pooled spleen and lymph nodes of C57/B6 mice (Harlan Laboratories) by magnetic bead selection (Miltenyi Biotec Inc., San Diego, CA). CD4+ cells were cocultured with CD4-depleted splenocytes that were irradiated with 3000 rads. T cells were activated with anti-CD3 (clone 145-2C11, generated at BMS) in presence of 5 ng/ml recombinant human TGF-β1 (R&D Systems) and 20 ng/ml recombinant mouse IL-6 (R&D Systems) along with 2 μg/ml of anti–IL-4 (clone 11B11; eBioscience) and 2 μg/ml anti–interferon-γ (anti–IFN-γ) (clone XMG1.2; eBioscience) neutralizing antibodies. After 6 days in culture, the polarized Th17 cells were harvested, reseeded in a 96-well Falcon plate and stimulated with 20 ng/ml rmIL-23 (R&D Systems) and 5 ng/ml rmIL-2 (eBioscience) along with 2 μg/ml of anti–IL-4 (clone 11B11; eBioscience) and 2 μg/ml anti–IFN-γ (clone XMG1.2; eBioscience) neutralizing antibodies for 4 days. The conditioned media from these cultures were harvested and assayed for both IL-17A and IL-22 concentrations by ELISA (R&D Systems). The IL-23–dependent response was measured by calculating the difference between the level of cytokine induced by the combination of rmIL-2 and rmIL-23 and the baseline level induced by rmIL-2 alone. To test the activity of ATI-1221 and ATI-1249 for inhibiting IL-23–dependent responses, a dose range of each inhibitor was added during restimulation of the Th17 cells with rmIL-2 and rmIL-23. Results are expressed as percent inhibition of IL-17A or IL-22 levels and IC50s were determined using XLfit software (ID Business Solutions Ltd., London, UK).
IL-17–Dependent CXCL1 Induction in NIH/3T3 Cells.
NIH/3T3 cells (CRL-1658; American Type Culture Collection) were seeded at concentration of 3 × 104 cells per well onto a 96-well flat-bottom Falcon plate. Cells were stimulated with 30 ng/ml recombinant mouse IL-17A/A (rmIL-17A/A) (generated at BMS) or 300 ng/ml recombinant mouse IL-17A/F (rmIL-17A/F) (R&D Systems) for 24 hours. Conditioned supernatant was harvested and tested for mouse CXCL1 concentrations by ELISA (R&D Systems). The IL-17–dependent response was evaluated by calculating the difference between the level of cytokine induced by rmIL-17A/A or rmIL-17A/F and the background level in cultures with media alone. To test the activity of mIL-17RA-Fc or the anti–IL-17 antibody for inhibiting IL-17–dependent responses, a dose range of each inhibitor was added during stimulation of the NIH/3T3 cells with rmIL-17A/A or rmIL-17A/F. Results are expressed as percent inhibition of CXCL1 levels and IC50s were determined using XLfit software.
Coculture of Th17 Cells with NIH/3T3 Cells.
Th17 cells were generated as described above and seeded at concentration of 2 × 105 cells per well onto a flat bottom plate along with 3 × 104 cells per well NIH/3T3 cells. Cultures were stimulated with 100 ng/ml rmIL-23 (R&D Systems) and 5 ng/ml rmIL-2 (eBioscience) along with 2 μg/ml of anti–IL-4 (clone 11B11; eBioscience) and 2 μg/ml anti–IFN-γ (clone XMG1.2; eBioscience) neutralizing antibodies for 4 days. The conditioned media from these cultures was harvested and assayed for IL-17A, IL-22, and IL-6 concentrations by ELISA (R&D Systems). To test the impact of ATI-1221 and mIL-17RA-Fc for inhibiting cytokine responses, each inhibitor at 20 nM concentration was added either alone or in combination during stimulation with rmIL-2 and rmIL-23. One-way analysis of variance (ANOVA) and Bonferroni’s post-test using GraphPad Prism software were performed on cytokine results to test for statistical significance of differences observed.
IL-23–Dependent In Vivo Pharmacodynamic Model.
Female C57BL/6 mice (Charles River Laboratories) at 8–9 weeks of age were acclimated for 3 days and were randomly assigned in treatment groups based on body weight. In this model, animals exposed to mIL-2 (eBioscience) and mIL-23 (generated at BMS) increased their serum levels of mIL-17 as a response from polyclonally activated T cells. A priming dose with mIL-2 was administered to all groups (except the control group) 24 hours before mIL-23 administration. When coadministered, mIL-2 and mIL-23 were diluted in PBS for a final 100 μl intraperitoneal administration per mouse. Two hours before the start of the challenge doses of mIL-2 plus mIL-23, a single dose of ATI-1249 formulated in PBS was administered subcutaneously at the doses indicated. Challenge doses of mIL-2 plus mIL-23 were given at 0, 7, and 23 hours. Optimal amounts of mIL-2, when coinjected with mIL-23, were determined experimentally and were used to maintain mIL-17 induction throughout the evaluation period. Thus, at time points 0 hours and 23 hours, 5 μg mIL-2 was used, whereas 10 μg was used at the 7-hour time point. A constant amount of 0.2 μg/mouse of mIL-23 was coadministered at all the indicated time points. After 30 hours the priming dose was experimentally determined to be the highest peak for mIL-17 induction in the absence of anti–IL-23 blockers. Thus, all treatment groups were anesthetized at 30 hours using 2.5% isoflurane inhalation and blood was terminally collected via cardiac puncture. Serum was separated by centrifugation, and serum samples were stored at –80°C analysis. Serum mIL-17 cytokine levels were evaluated by ELISA (R&D Systems), following the manufacturer’s instructions. Serum mIL-22 cytokine levels were evaluated using EMD Millipore (Billerica, MA) Luminex Milliplex kit, following the manufacturer’s instructions. One way ANOVA and Dunnett’s multiple comparison tests using GraphPad Prism software were performed on cytokine results to test for statistical significance of observed differences between treatments.
IL-17–Dependent In Vivo Pharmacodynamic Model.
Female BALB/c mice (Harlan Laboratories) at 8–10 weeks of age were dosed subcutaneously with the anti–IL-17 antibody at various doses. Eighteen hours later, mice were challenged with rmIL-17A/A (150 μg/kg) (generated at BMS) or rmIL-17A/F (150 μg/kg) (R&D Systems) subcutaneously in 0.2 ml of PBS. Two hours later, serum samples were collected and the levels of CXCL1 were measured by ELISA (R&D Systems). One-way ANOVA and Dunnett’s multiple comparison tests using GraphPad Prism software were performed on cytokine results to test for statistical significance of the differences observed between treatment.
Mouse Imiquimod-Induced Skin Inflammation Model.
Female BALB/c mice (Harlan Laboratories) at 6–9 weeks of age were randomly assigned to treatment groups (n = 8). Three days before initiation of the study, mice were shaved along their backs (from shoulder to hip) with electric clippers, and a depilation lotion (Nair; Church & Dwight Co., Ewing, NJ) was applied for complete removal of hair. Anesthetized mice (2.5% isoflurane inhalation) received a dose of 62.5 mg/mouse of imiquimod (IMQ Cream 5%; Graceway Pharmaceuticals, Bristol, TN) applied with a wooden applicator to the shaved skin. In the prophylactic study design beginning 18 hours before IMQ treatment, mice received either PBS, anti–IL-17 antibody (20 mg/kg s.c.), ATI-1249 (20 mg/kg s.c.), or a combination of the two therapeutics. Skin thickness was measured daily using a Mitutoyo dial caliper (model no. 2412F; Mitutoyo, Kawasaki, Japan). The dorsal skin was visually assessed for erythema and scaling daily using the following criteria: 0, none; 1, slight; 2, moderate; 3, marked; 4, severe. Skin thickness was calculated as the percent change in thickness from baseline measurement taken before the start of IMQ application for each animal. The results are expressed as mean ± S.E.M. for each group. Data calculations were performed in Microsoft Excel (Redmond, WA), while statistical analysis (one-way ANOVA with Bonferroni’s post-test) and graphics were performed using GraphPad Prism software. After seven applications of IMQ using 2.5% isoflurane inhalation, all treatment groups were anesthetized and blood was collected via cardiac puncture. Serum was separated by centrifugation and samples were stored at –80°C until analysis. Serum cytokine levels were evaluated using Luminex multiplex kits (EMD Millipore) following the manufacturer’s instructions. One-way ANOVA and Bonferroni’s post-test using GraphPad Prism software was performed on cytokine results to test for statistical significance of differences observed between treatment groups.
Histology for Mouse IMQ-Induced Skin Inflammation Model.
Dorsal skin from five animals per group was collected at necropsy, fixed in 10% neutral buffered formalin, routinely processed to paraffin, and sectioned at 5 μm for H&E evaluation of lesion severity. To score the severity of inflammation of the dorsal skin, an objective scoring system was developed based on the following parameters: extent of the lesion, the severity of hyperkeratosis, number and size of pustules, height of epidermal hyperplasia (acanthosis, measured in interfollicular epidermis), and the amount of inflammatory infiltrate in the dermis and soft tissue. Acanthosis and inflammatory infiltrate were scored independently on a scale from 0 to 4: 0, none; 1, minimal; 2, mild; 3, moderate; 4, marked.
Mouse Experimental Autoimmune Encephalomyelitis Model.
C57BL/6 female mice at 6–8 weeks of age (The Jackson Laboratories) were immunized subcutaneously with a total of 200 μl over two areas with 3 mg/ml MOG35–55 emulsified 1:1 with 3 mg/ml Mycobacterium tuberculosis (Difco Laboratories/Becton Dickinson, Franklin Lakes, NJ) in Incomplete Freund’s Adjuvant (Sigma-Aldrich) (300 μg peptide and 300 μg M. tuberculosis per mouse). Two hours later, mice were injected intraperitoneally with 300 ng pertussis toxin (in 100 μl volume). One day before immunization, mice were treated subcutaneously with PBS (every other day), the anti–IL-23 adnectin, ATI-1249 (25 mg/kg every other day), the anti–IL-17 antibody (25 mg/kg 1×/week), or combination of ATI-1249 and the anti–IL-17 antibody with the same regimen used in the monotherapy groups. Clinical scoring and body weight were taken three times per week. Clinical scoring system: 0.5, partial tail weakness; 1, limp tail or waddling gait with tail tonicity; 1.5, waddling gait with partial tail weakness; 2, waddling gait with limp tail (ataxia); 2.5, ataxia with partial limb paralysis; 3, full paralysis of one limb; 3.5, full paralysis of one limb with partial paralysis of a second limb; 4, full paralysis of two limbs; 4.5, moribund; 5, death. Mean clinical score was calculated by averaging the scores of all mice in each group. One-way ANOVA with Bonferroni’s post-test were performed using GraphPad Prism software on clinical score data to test for statistical significance of the differences observed between treatment groups.
Histology for Mouse EAE Model.
At the end of study, intact spinal columns were removed and fixed in 10% neutral buffered formalin. Three transverse sections of the lumbar segment of spinal cord were excised and routinely processed to paraffin. Paraffin-embedded tissues were sectioned at 3 μm for H&E, and 6 μm for the myelin-specific stain Luxol fast blue. Slides were analyzed in a blinded fashion for severity of inflammation and demyelination. A histologic score of 0 to 5 was assigned separately for each lesion characteristic for all three transverse sections of spinal cord evaluated per animal. A score of 0 indicates no lesions/infiltrate; 1, minimal; 2, mild; 3, moderate; 4, marked; and 5, severe. Average scores of inflammation and demyelination were calculated and reported.
Results
The Anti-Murine IL-23–Specific Adnectins ATI-1221 and ATI-1249 Potently Inhibit IL-23–Dependent Responses In Vitro.
To selectively and specifically neutralize murine IL-23, a murine IL-23p19–specific adnectin was generated using mRNA display (Roberts and Szostak, 1997). An adnectin is a recombinant protein derived from the tenth type III domain of fibronectin that has been modified to bind specifically to a selected target. The functional potency of the adnectins was evaluated for inhibition of murine IL-23–dependent pSTAT3 signaling in the human cell line Kit225. As a result of the 70% homology between mouse and human IL-23, stimulation of Kit225 cells with murine IL-23 induced a robust 8- to 9-fold induction of pSTAT3 above background (Oppmann et al., 2000). Using this assay we obtained a series of mIL-23 binding adnectins. To improve the affinity of these molecules against mIL-23, an optimization-selection was carried out using increased selective pressure, including lowering target concentration. From this optimization process, ATI-1221 was identified as a potent IL-23–specific inhibitor with subnanomolar binding affinity to IL-23 (Supplemental Table 1). In the Kit225 assay, ATI-1221 dose dependently inhibited the IL-23–induced activation of pSTAT3 with an IC50 of 1.5 nM (Fig. 1, A and B).

Mouse anti–IL-23 adnectins ATI-1221 and ATI-1249 potently inhibit IL-23–dependent responses in vitro. (A) Average IC50 values calculated from at least four experiments for anti–IL-23 adnectins in the IL-23–dependent pSTAT3 induction assay in Kit225 cells and the IL-23–dependent cytokine secretion assay in polarized Th17 cells. (B) IC50 curves shown were generated from one study and are representative of at least four replicate studies testing the inhibition of IL-23–induced pSTAT3 by ATI1221 and ATI-1249 in the IL-23–dependent pSTAT3 induction assay in Kit225 cells. Data points are the percent inhibition calculated from the average value of duplicate wells. Error bars represent S.E.M. (C and D) IC50 curves shown were generated for one study and are representative of least four replicate studies testing the inhibition of IL-23–dependent IL-17A (C) and IL-22 (D) by ATI1221 or ATI-1249 in the IL-23–dependent cytokine secretion assay in polarized Th17 cells. For (C) and (D) data points are the percent inhibition calculated from the average value of triplicate wells.
To test ATI-1221 for activity against IL-23–dependent responses in primary cells, an assay was established for inducing IL-23–driven cytokine secretion from differentiated mouse Th17 cells. T cells were polarized to a Th17 phenotype by incubation with TGF-β1 and IL-6 for 6 days. The Th17 cells were harvested and recultured with IL-2 and IL-23 to induce IL-17A and IL-22. The addition of IL-2 was required to maintain cell viability and enable robust cytokine production but did not strongly induce IL-17A or IL-22 by itself. Addition of IL-23 to IL-2 typically caused a 2- to 3-fold induction of IL-17A and at least a 15-fold induction of IL-22. Addition of a range of concentrations of ATI-1221 during stimulation of Th17 cells with IL-2 and IL-23 caused potent inhibition of both IL-23–dependent IL-17A and IL-22, with comparable IC50 values of 1.6 and 1.9 nM, respectively (Fig. 1, A, C, and D).
To increase the pharmacokinetic properties of ATI-1221, the adnectin was reformatted as a PEGylated molecule and renamed ATI-1249. To ensure that PEGylation did not affect potency, it was tested for functional activity in vitro in the IL-23–induced pSTAT3 assay in Kit-225 cells and also the primary Th17 cell assay. Similar to the unformatted adnectin, ATI-1249 bound IL-23 with subnanomolar affinity and inhibited pSTAT3 in a dose-dependent manner with an IC50 of 2.6 nM (Supplemental Table 1; Fig. 1, A and B). In the primary Th17 cell assay, ATI-1249 also inhibited IL-23–dependent cytokine secretion from Th17 cells with an IC50 of 1.9 nM for inhibition of IL-17A and 2.3 nM for inhibition of IL-22 (Fig. 1, A, C, and D). Therefore, PEGylation of ATI-1249 did not adversely affect the potency of the anti–IL-23 adnectin. The specificity of ATI-1221 and ATI-1249 for IL-23 over IL-12 was confirmed by demonstrating that ATI-1221 and ATI-1249 had no effect on IL-12–dependent mouse Th1 differentiation and secretion of IFN-γ compared with an anti-p40 antibody (Supplemental Fig. 1). Together these data established ATI-1221 and ATI-1249 as potent and selective inhibitors of IL-23–dependent responses in vitro.
The mIL-17RA-Fc and Anti-Mouse IL-17 Antibody Potently Inhibit IL-17–Dependent CXCL1 In Vitro.
IL-17 acts on various cell types, including epithelial cells, endothelial cells, and fibroblasts to induce numerous cytokines and chemokines, such as IL-6, granulocyte colony-stimulating factor, GM-CSF, IL-1β, TGF-β, tumor necrosis factor-α, IL-8, and CXCL1 (Aggarwal and Gurney, 2002; Weaver et al., 2007). To test the impact of IL-17 neutralization on cellular responses in vitro we generated a soluble decoy receptor mouse IL-17RA-Fc as well as a monoclonal antibody against mouse IL-17. To measure the activity and selectivity of mIL-17RA-Fc and the anti-mouse IL-17 antibody for inhibition of IL-17–dependent responses, these agents were tested for inhibition of murine IL-17A/A and IL-17A/F–dependent induction of CXCL1 from NIH/3T3 fibroblast cells. Stimulation of NIH/3T3 cells for 24 hours with mouse IL-17A/A or IL-17A/F elicited a greater than 15- or 200-fold induction of CXCL1, respectively. Treatment with mIL-17RA-Fc caused a dose-dependent inhibition of IL-17A/A–induced activity with an IC50 of 1.8 nM (Fig. 2, A and B), and IL-17A/F–induced activity with an IC50 of 2.6 nM (Fig. 2, A and C). The anti–IL-17 antibody was slightly more potent against IL-17A and IL-17A/F with an IC50 of 0.9 nM and 1.2 nM, respectively (Fig. 2). These data confirmed that both the mIL-17RA-Fc and the anti–IL-17 antibody were potent and selective inhibitors of mouse IL-17A/A and IL-17A/F in vitro.

Mouse anti–IL-17 inhibitors, mIL-17RA-Fc and anti–IL-17 Ab, potently inhibit IL-17–dependet responses in vitro. (A) Average IC50 values calculated from at least three experiments for anti–IL-17 inhibitors against IL-17A/A and IL-17A/F–dependent induction of CXCL1 from NIH/3T3 fibroblast cells. (B) IC50 curves shown were generated from one study and are representative of at least three replicate studies testing the inhibition of IL-17A/A–induced or (C) IL-17A/F–induced CXCL1 by mIL-17RA-Fc and anti–IL-17 in NIH/3T3 cells stimulated for 24 hours. Each data point represents the percent inhibition calculated from the average CXCL1 concentration in duplicate wells.
Dual Inhibition of IL-23 and IL-17 In Vitro Offers More Complete Suppression of Cytokine Secretion.
To extend our findings with IL-23 and IL-17 inhibitors in distinct IL-23– and IL-17–dependent assays, we sought to test the impact of dual inhibition of IL-23– and IL-17–dependent responses within the same assay system. To test for potential cooperative effects of inhibiting both IL-23 and IL-17 in vitro, a method was developed in which polarized mouse Th17 cells were cocultured with NIH/3T3 cells. In this system, Th17 cells served as the IL-23–responsive population and the source of IL-17, and the NIH/3T3 cells were the IL-17–responsive cells. Given a role for IL-6 more physiologically relevant than CXCL1 in the development of Th17-mediated pathology, IL-6 was used as the primary readout of IL-17–dependent activity in the NIH/3T3 cells (Yao et al., 1995; Hwang et al., 2004; Yen et al., 2006; Chiricozzi and Krueger, 2013). Addition of IL-23 induced IL-17A production from the Th17 cells, which in turn had a downstream effect of driving cytokines from NIH/3T3 cells, including IL-6 (Fig. 3, A and C). Notably, in parallel to IL-17 production, IL-23 could also drive IL-22 production from Th17 cells, providing an additional readout of IL-23 function (Fig. 3B). Using this method, the impact on cytokine production of blocking IL-23 and IL-17 alone or in combination was tested. A dose of the anti–IL-23 adnectin ATI-1221 and the mIL-17RA-Fc fusion was chosen on the basis of the potency in the initial functional in vitro assessment such that saturating levels of each agent were used corresponding to approximately 10-fold over the IC50 in the primary cell–based assays described above.

Dual inhibition of IL-23 and IL-17 in a coculture of Th17 and NIH/3T3 cells is more effective for inhibiting cytokine production than either alone. ELISA measurements of IL-17A (A), IL-22 (B), and IL-6 (C) concentrations in conditioned supernatants collected from cocultures of polarized Th17 cells with NIH/3T3 cells and stimulated with IL-2 alone or IL-2+IL-23 for 4 days. The impact of IL-23 and IL-17 inhibition on cytokine levels was tested by adding media alone (open bars), 20 nM ATI-1221 alone (gray bars), 20 nM mIL-17RA-Fc alone (hatched bars), or both inhibitors combined (solid bars). Graphs show data from one study and are representative of at least three comparable studies. Data are calculated as the average cytokine concentration from triplicate wells. Error bars represent S.E.M. *P < 0.01 by one-way ANOVA with Bonferroni’s post-test for multiple comparisons for IL-2 versus IL-2+IL-23 stimulation, (#) for media versus any treatment, and (^) for single inhibitor versus combination treatment. Note: All cytokine levels elevated by IL-2 or IL-2+IL-23 regardless of treatment were statistically significantly different compared with the media alone condition.
In this coculture system, stimulation with IL-2 alone led to an increase in IL-17A and IL-22 consistent with the ability of IL-2 to act as a T-cell survival and proliferation factor promoting increased levels of T-cell cytokines in the culture. Importantly, the low levels of IL-6 produced by the coculture system were also increased with the addition of IL-2, consistent with the increases in T-cell cytokines in this system. Of note, blockade of IL-23 did not affect the modestly increased IL-17A– or IL-22–cytokine production driven by IL-2 alone, suggesting that production of these cytokines was not dependent on IL-23 (Fig. 3, A and B). To further activate the IL-23–IL-17 pathway, IL-23 was added to the coculture system and dramatically increased IL-17A as well as IL-22 production, consistent with the important role of IL-23 in directly driving these cytokines from T cells (Fig. 3A) (Liang et al., 2006). Blockade of IL-23 with the anti–IL-23 adnectin ATI-1221 alone caused a greater than 95% inhibition of IL-17A (Fig. 3A) and 92% inhibition of IL-22 (Fig. 3B). Production of IL-22 driven either by IL-2 or IL-2/IL-23 was not affected by the inhibition of IL-17 and was blocked similarly by either the IL-23 adnectin or the combination of the IL-23 adnectin with mIL-17RA-Fc (Fig. 3B). These data suggest minimal or no contribution of the IL-17 neutralization in the combination treatment, which might have been expected considering the exclusive dependence of IL-22 on IL-23 (Liang et al., 2006).
In addition, IL-6 was detected in these cultures at baseline (media alone) and was further increased by the addition of IL-2 or IL-2/IL-23 (Fig. 3C). IL-23 blockade had no effect on IL-6 in the baseline or IL-2-stimulated cultures. The IL-6 levels in cultures with IL-2 alone was modestly but significantly augmented approximately 25% by the addition of IL-23 (Fig. 3C). While the IL-6 levels seem to be very dependent on IL-17 levels detected in the culture, the dramatically increased IL-17 produced in the presence of IL-23 caused only a modest enhancement of IL-6 production from the NIH/3T3 cells. Consistent with this notion, IL-23 neutralization had only a modest effect for blocking IL-6 driven by IL-2/IL-23 but did reduce the IL-6 levels to the level induced by IL-2 alone. However, the amount of IL-6 in the culture was still markedly elevated in the presence of IL-23 blockade. Notably, IL-17 production was not completely eliminated in the presence of IL-23 neutralization (Fig. 3A). The level of IL-17 that persisted was actually comparable to the amount observed with IL-2 alone, suggesting that IL-17 was produced independently of IL-23. Clearly, this amount of IL-17 was sufficient to drive robust IL-6 secretion even in the presence of IL-23 neutralization. However, the IL-6 production under all conditions, including baseline, was inhibited by IL-17 blockade, consistent with the hypothesis that IL-6 production in the NIH/3T3 cells was driven by IL-17 expressed either at baseline or further induced by IL-2 and IL-2/IL-23, with an 85% inhibition of IL-6 at baseline, 67% inhibition of IL-6 after IL-2 treatment alone, and 55% inhibition in the IL-2 /IL-23 costimulation observed (Fig. 3C). Of note, neutralization of IL-17, however, did not block the levels of IL-6 in the presence of IL-2/IL-23 to the level observed with IL-2 alone. These data suggest that the IL-17 inhibition alone was insufficient to reduce the IL-6 levels that were augmented with IL-23. Together, these results demonstrate that IL-23 blockade alone was ineffective for complete inhibition of IL-17–dependent IL-6 production, and IL-17 blockade alone was also inadequate for total inhibition of IL-23–dependent responses, including production of IL-22.
When both ATI-1221 and mIL-17RA-Fc were added together in cultures stimulated with IL-23 and IL-2, the dual inhibition further reduced IL-6 levels more than 69% (Fig. 3C). This reduction of IL-6 was 37% lower than the level observed in the presence of IL-17 blockade alone and was comparable to the level induced by IL-2 alone (Fig. 3C). The augmented suppression of IL-6 levels suggest that the IL-23 inhibition abolished the IL-23–dependent IL-17 as observed with ATI-1221 alone, and the mIL-17-Fc further blocked any residual IL-17 secreted independently of IL-23. This comprehensive neutralization of IL-17 with dual inhibition led to the more profound reduction of IL-6. Despite the fact that the IL-23 adnectin was virtually ineffective for reducing IL-6 levels on its own, a reduction was observed when it was combined with IL-17 neutralization, indicating that better blockade of the IL-17–IL-23 axis could be achieved by dual inhibition. Collectively, these data from the coculture system demonstrate that dual targeting of both IL-23 and IL-17 not only completely inhibited IL-23–dependent IL-22 but also had a more dramatic effect by blocking IL-6 levels as well.
The PEGylated Anti-Murine IL-23–Specific Adnectin ATI-1249 Inhibits IL-23–Dependent Activity In Vivo.
With the aim of ultimately testing the impact of IL-23 and IL-17 dual inhibition in vivo, we validated our inhibitors in IL-23– and IL-17–dependent pharmacodynamic (PD) models. First, we evaluated the activity of the PEGylated anti–IL-23 adnectin ATI-1249 for inhibiting IL-23–dependent responses in vivo. The PD model used to test the functional activity of ATI-1249 against IL-23–dependent responses in vivo involved administering mIL-2 and mIL-23 to mice according to a specific regimen (Fig. 4A). IL-2 was injected alone 24 hours before three consecutive treatments with IL-2 and IL-23 at 0, 7, and 23 hours. Three challenges of IL-2/IL-23 were required to detect robust cytokine levels in the serum. At 30 hours, the mice were bled and serum was processed for measuring IL-17 and IL-22 concentrations by ELISA. Challenge with IL-2 alone or mouse IL-23 alone had minimal effects on cytokine secretion, but the combination of IL-23 with IL-2 resulted in robust production of IL-17A and IL-22 (Fig. 4, B and C). To test the activity of ATI-1249 for inhibition of these IL-23–dependent cytokines, mice were treated with a range of ATI-1249 concentrations 2 hours before the first challenge with IL-2/IL-23 (Fig. 4A). In these mice, a dose response was observed, with maximal reduction of both IL-17 and IL-22 obtained at 5 mg/kg of ATI-1249, corresponding to 98 and 88% reductions, respectively (Fig. 4, B and C). These data demonstrate that ATI-1249 caused potent inhibition of IL-23–dependent cytokine responses in vivo and suggest that good pharmacodynamic coverage of IL-23 was obtained during the 32 hours of the experiment post-adnectin dosing.

Mouse anti–IL-23 adnectin ATI-1249 inhibits IL-23–dependent cytokines in vivo. (A) Experimental design of the IL-23–dependent PD assay, (B) ELISA measurements of IL-17A, and (C) IL-22 in serum collected from mice challenged by intraperitoneal injection with one dose of IL-2 alone and three subsequent doses of IL-2+IL-23 at 24, 31, and 47 hours after the first treatment with IL-2. To test the impact of IL-23 inhibition, mice were dosed by subcutaneous injection with a range of ATI-1249 concentrations 2 hours before the first challenge with IL-2+IL-23. A terminal bleed was performed and serum was processed for analysis at 56 hours. The results are expressed as the mean IL-17 or IL-22 level in serum measured in five mice per group. Error bars represent S.E.M. #P < 0.05 by one-way ANOVA with Dunnett’s multiple comparison test for IL-2+IL-23 versus either IL-2+PBS or the IL-23+PBS controls. *P < 0.05 by one-way ANOVA with Dunnett’s multiple comparison test for ATI-1249–treated groups versus untreated IL-2+IL-23 control.
The Mouse Anti–IL-17 Antibody Inhibits IL-17A/A– and IL-17A/F–Dependent Activity In Vivo.
Next, we tested our anti–IL-17 inhibitors for activity in vivo. Because the anti–IL-17 antibody was slightly more potent against IL-17A/A and IL-17A/F activity than the mIL-17-RA decoy receptor in vitro, we tested the activity of anti–IL-17 antibody for inhibition of IL-17A/A and IL-17A/F responses in vivo. The PD model established for investigating IL-17–dependent responses in vivo involved subcutaneous challenge of mice with mouse IL-17A/A or IL-17A/F followed by blood collection at 2 hours after treatment. Serum was subsequently processed and tested for CXCL1 levels by ELISA. Mice challenged with either IL-17A/A or IL-17A/F exhibited a robust secretion of CXCL1 into the serum (Fig. 5, A and B). Administering a range of anti–IL-17 antibody concentrations 18 hours before either IL-17A/A or IL-17A/F challenge caused significant inhibition of CXCL1. With IL-17A/A challenge, the anti–IL-17 antibody caused a dose-dependent inhibition of CXCL1, in which the highest dose tested, 1.0 mg/kg, blocked 92% of the untreated control (Fig. 5A). The anti–IL-17 antibody was less potent in blocking IL-17A/F–induced CXCL1 in vivo but still significantly inhibited secretion at the highest doses tested, both the 0.2 and 1.0 mg/kg doses showing approximately 50% inhibition of CXCL1 production (Fig. 5B). These data demonstrated that the anti–IL-17 antibody had potent activity against both IL-17A/A and IL-17A/F activity in vivo, with good pharmacodynamic coverage through the 20 hours of the experiment.
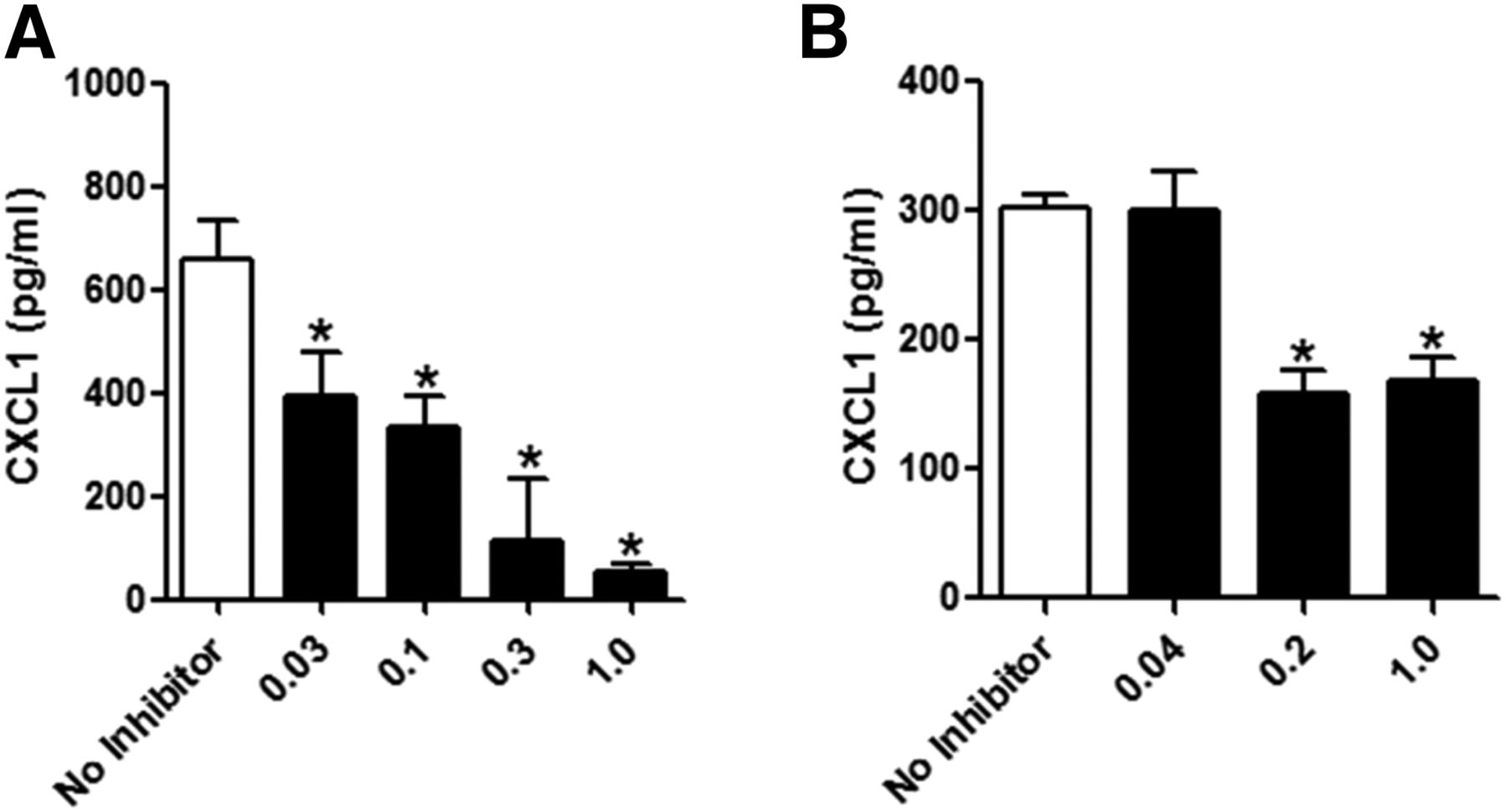
Mouse anti–IL-17 antibody inhibits IL-17–dependent responses in vivo. ELISA measurements of CXCL1 in serum from mice challenged subcutaneously with (A) IL-17A/A or (B) IL-17A/F. To test the impact of IL-17 inhibition, mice were dosed with anti–IL-17 antibody by subcutaneous injection 18 hours before challenge with IL-17A/A or IL-17A/F. Two hours after challenge, serum was collected for analysis. The results are expressed as mean CXCL1 level in serum measured in four mice per cohort. Error bars represent S.E.M. *P < 0.05 by one-way ANOVA with Dunnett’s multiple comparison test for anti–IL-17 antibody treatment versus “no inhibitor,” which for (A) was mIgG isotype control and for (B) was PBS.
Dual Inhibition of Mouse IL-23 and IL-17 Offers Superior Efficacy in the Imiquimod-Induced Skin Inflammation Model.
Treatment of mouse skin with IMQ leads to skin inflammation, including increases in skin thickness and scaling, that has been reported to be dependent on IL-17 and IL-23 on the basis of the protection from these symptoms in mice deficient for IL-23p19 and IL-17 receptor (van der Fits et al., 2009). Therefore, we tested whether inhibition of both IL-23 and IL-17 might offer superior efficacy compared with inhibition of either alone in the IMQ skin inflammation model. In this model, IMQ was applied to the skin on backs of shaved mice for 7 consecutive days. Successive IMQ treatments caused a rapid increase in skin thickness in the first 4 days with a greater than 70% change from baseline at day 4 (Fig. 6A). Skin scaling also increased, reaching a score of 3 by day 7 (Fig. 6B). Histologic examination of the skin showed that there were minimal-to-moderate epidermal hyperplasia (acanthosis) and moderate-to-marked mixed-cell infiltrates of the dermis in the back skin of IMQ-induced mice (Fig. 6, C, G, and H).

Dual inhibition of IL-23 and IL-17 offers superior efficacy in the IMQ-induced skin inflammation model. Three days before IMQ challenge mice were anesthetized with isoflurane and their back hair was removed. One day before IMQ application and every other day thereafter animals were dosed subcutaneously with PBS, 20 mpk ATI-1249 alone, 20 mpk anti–IL-17 antibody alone, or both combined. Imiquimod was applied every day from days 0 to 6. (A) Skin thickness and (B) scaling was measured daily. Results are expressed as the mean values for eight animals per cohort. Error bars represent S.E.M. *P < 0.05 by one-way ANOVA with Bonferroni’s post-test for multiple comparisons for any treatment versus vehicle, (#) anti–IL-17 antibody versus ATI-1249, or (+) anti–IL-17 antibody versus the combination treatment. ***P < 0.001 by one-way ANOVA with Bonferroni’s post-test for multiple comparisons for the combination treatment versus vehicle. At necropsy, back skin was prepared for histology and analyzed for acanthosis, and dermal infiltrate by neutrophils, eosinophils, plasma cells, lymphocytes, and melanocytes. Representative images were scored as follows: (C) PBS–acanthosis, 3; dermal infiltrate, 4; (D) ATI-1249–acanthosis, 2; dermal infiltrate, 2; (E) anti–IL-17 Ab–acanthosis, 2; dermal infiltrate, 3; (F) combination–acanthosis: 1; dermal infiltrate, 1. From five animals in each treatment group the average histology score was calculated for (G) acanthosis and (H) dermal infiltrate. Error bars represent S.E.M. *P < 0.01 between the combination treatment versus PBS by Kruskal-Wallis test with Dunn’s post-test for multiple comparisons.
Preventative dosing of both inhibitors in this model was performed at doses expected to give robust suppression of the target cytokines. On the basis of the nearly maximal activity of this antibody at 1 mg/kg in the IL-17–dependent PD model, complete neutralization of IL-17A would be expected at the high dose (20 mg/kg) used in this model. Similarly, neutralization of IL-23 by 20 mg/kg ATI-1249 would be expected on the basis of the near maximal suppression of IL-23–mediated effects at 5 mg/kg in the IL-23–dependent PD model described earlier. Treatment with the anti–IL-17 antibody 1 day before IMQ application and every other day thereafter had no significant effect on preventing skin thickening or scaling (Fig. 6, A and B). In contrast, treatment with a similar regimen of ATI-1249 caused reduced thickness starting at day 5 and was 38% lower than the vehicle control at day 7 but did not reach statistical significance compared with the vehicle control. However, ATI-1249 did have a significant effect for reducing skin scaling by day 6 (Fig. 6B). Dosed simultaneously, the combination of ATI-1249 and anti–IL-17 antibody caused an effect similar to that of ATI-1249 alone for reducing skin thickening initially and by day 7 had a superior effect. Notably, only the dual treatment induced a statistically significant reduction in skin thickening compared with the vehicle control. A more dramatic effect of the combination dosing was observed on the skin scaling score where no increase in scaling was observed beyond 4 days with combination treatment. Moreover, although treatment with ATI-1249 alone and together with anti–IL-17 antibody caused significant reductions in the scaling score, the combination therapy was still comparatively superior (Fig. 6B). The suboptimal efficacy of the monotherapies and the superiority of the combination treatment were further supported by histologic evaluation of skin collected from treated mice (Fig. 6, C–F). In the presence ATI-1249 or the anti–IL-17 antibody alone, there was no significant effect on the acanthosis score. The combination treatment, on the other hand, reduced acanthosis 55% compared with the control or either of the monotherapies but did not reach statistical significance (Fig. 6G). For the dermal infiltrate analysis, the group mean scores were reduced 31% by ATI-1249 alone and 38% by anti–IL-17 antibody alone compared with the vehicle control group (Fig. 6H). With the combination treatment, the infiltrates were significantly reduced by 69% compared with vehicle and over 50% compared with either ATI-1249 or anti–IL-17 antibody alone.
Analysis of serum cytokines in IMQ challenged mice showed that several IL-23 and IL-17–associated cytokines were increased, including IL-17A, IL-22, and IL-6 (Fig. 7, A–C). However, owing to the local nature of the inflammation in the skin in this model, the overall levels of the cytokines remained low and exhibited considerable interanimal variability. Nonetheless, a reduction of cytokine levels was observed in the serum from treated animals, which were in line with expectations from the in vitro experiments. In mice treated with ATI-1249 alone, the group mean concentration for IL-17A and IL-22 but not IL-6 was reduced. The anti–IL-17 antibody did not reduce IL-22 or IL-6. However, treatment with the combination of ATI-1249 and the anti–IL-17 antibody caused decreases in both IL-22 and IL-6 serum levels (Fig. 7, B and C). These effects on serum cytokines in this in vivo model corroborate the cooperative effects of the combination treatment observed in vitro. Collectively, data from the analysis of the skin thickness, scaling, histology, and serum cytokine levels all suggest that dual inhibition of IL-23 and IL-17 in the IMQ skin inflammation model offered superior efficacy compared with neutralization of either IL-23 or IL-17 alone.
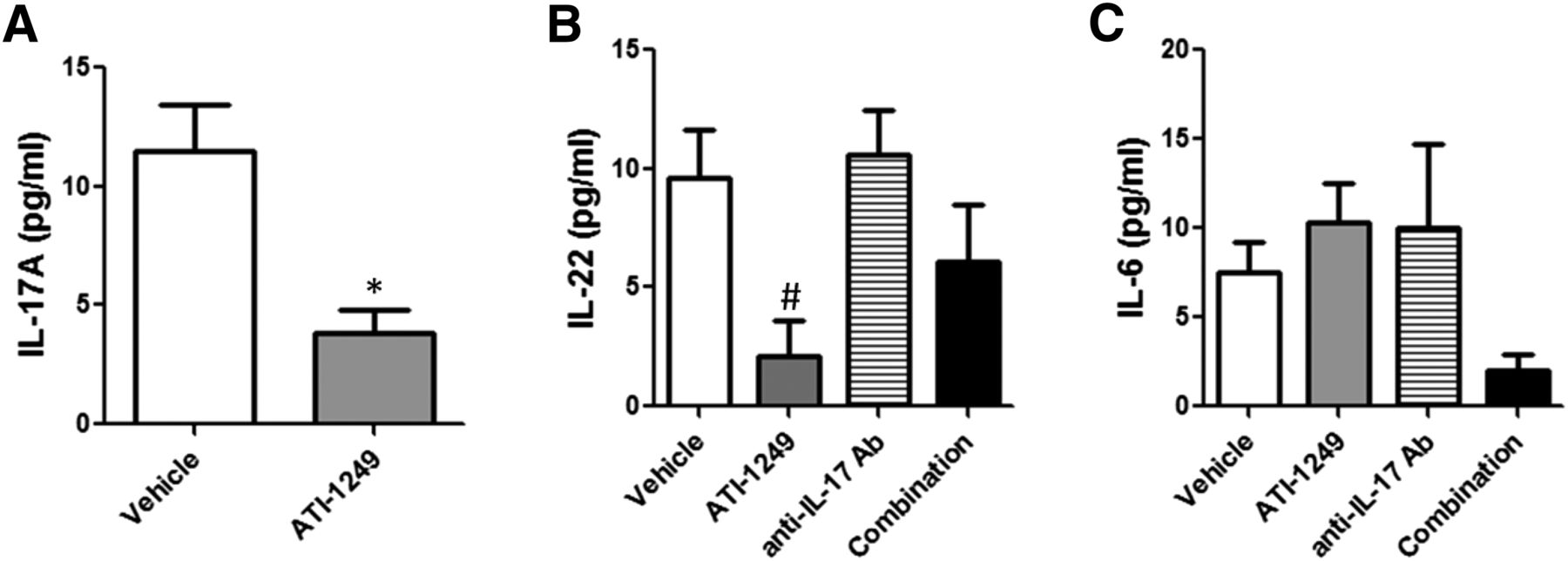
Dual inhibition of IL-23 and IL-17 enhances inhibition of serum cytokines in the IMQ-induced skin inflammation model. Serum was analyzed by ELISA for (A) IL-17A, (B) IL-22, and (C) IL-6 concentrations. Data are presented as the mean cytokine concentration from 10 animals per cohort in the presence of vehicle (open bars), ATI-1249 alone (gray bars), anti–IL-17 alone (hatched bars), or both inhibitors combined (solid bars). Error bars represent S.E.M. *P < 0.05 by one-way ANOVA with Bonferroni’s post-test for multiple comparisons for any treatment versus vehicle or (#) anti–IL-17 antibody versus ATI-1249.
Dual Inhibition of Mouse IL-23 and IL-17 Offers Superior Efficacy in the MOG Peptide-Induced EAE Model.
Similar to the IMQ-induced skin inflammation model, the IL-23–IL-17 axis has been shown to be central to inhibition of disease development in models of central nervous system (CNS) inflammation (Hofstetter et al., 2005; Langrish et al., 2005; Chen et al., 2006; Kroenke et al., 2008; Ishigame et al., 2009; Hu et al., 2010; El-Behi et al., 2011; Kap et al., 2011). To test whether dual inhibition of IL-23 and IL-17 offers superior efficacy compared with either inhibitor alone for treatment of CNS inflammation, we tested ATI-1249 and the anti–IL-17 antibody dosed alone or in combination in a MOG peptide–induced EAE model. In this model, 25 mg/kg of each agent was used. On the basis of IL-23 and IL-17 challenge models, the dose of inhibitors used was 5- and 25-fold over the maximal efficacious dose for good pharmacodynamic coverage in mice, respectively.
Induction of EAE in control animals resulted in rapid weight loss starting at day 9 along with onset of clinical symptoms that reached maximum severity by 12–14 days (Fig. 8, A and B). Treatment with ATI-1249 or the anti–IL-17 antibody alone administered just before disease onset at day 7 caused a modest reduction in disease score compared to vehicle-treated animals. Either inhibitor dosed by itself also had a modest effect for preventing weight loss (Fig. 8, A and B). Whereas the protection by the anti–IL-17 antibody was maintained to day 20, protection waned in mice treated with ATI-1249 alone. When both ATI-1249 and the anti–IL-17 antibody were administered together at day 7, the combination treatment caused a more dramatic reduction in the severity of clinical disease and prevented weight loss more significantly than either treatment alone (Fig. 8, A and B).

Dual inhibition of IL-23 and IL-17 offers superior efficacy in the MOG peptide–induced EAE model. Mice were immunized with MOG peptide emulsified with incomplete Freund's adjuvant and two injections of pertussis toxin. Mice were treated subcutaneously with PBS (every other day), ATI-1249 (25 mg/kg every other day), anti–IL-17 antibody (25 mg/kg once per week), or combination of ATI-1249 and anti–IL-17 antibody (the same regimen used in the monotherapy groups). Dosing was initiated the day before immunization. Superior efficacy is demonstrated with the combination of ATI-1249 and the anti–IL-17 antibody based on (A) clinical scores and (B) body weight. Results are expressed as the mean values for 14 animals per cohort. Error bars represent S.E.M. *P < 0.05 by one-way ANOVA with Bonferroni post-test for multiple comparisons for any treatment versus vehicle, (#) anti–IL-17 antibody versus ATI-1249 (^) ATI-1249 versus the combination treatment or (+) anti–IL-17 antibody versus the combination. Representative images are transverse sections of lumbar spinal cord from mice induced with MOG35–55 EAE, stained with luxol fast blue, and H&E counterstain to demonstrate reduction in inflammation (arrow), active demyelinating lesions (arrow head), and tissue damage (*) comparing treatment with (C) PBS, (D) ATI-1249, (E) anti–IL-17 Ab, and (F) combination therapy. Scale bar, 100 μm. A cumulative score was calculated for (G) inflammation and (H) demyelination for each treatment group. Results are expressed as the mean values for seven animals per cohort. Error bars represent S.E.M. *P < 0.01 between combination treatment compared with PBS by Kruskal-Wallis test with Dunn post-test for multiple comparisons.
Histologic examination of the spinal cords from EAE mice treated with ATI-1249 and the anti–IL-17 antibody alone or in combination corroborated the superiority of the dual treatment for preventing disease (Fig. 8, C–F). Dosed alone, ATI-1249 or the anti–IL-17 antibody provided partial protection on the basis of inflammation and demyelination scores (Fig. 8, G and H). In contrast, the combination of ATI-1249 or the anti–IL-17 antibody yielded a more significant effect in reduction of spinal cord inflammation and demyelination scores (Fig. 8, G and H). These data demonstrated that in the MOG peptide–induced EAE model the dual inhibition of IL-23 and IL-17 offered a superior effect compared with either inhibitor alone for reduction of overall disease severity by all measures: clinical scores, body weight loss, spinal cord inflammation, and demyelination.
Discussion
The generation of potent and specific neutralizing molecules against mouse IL-23p19 and IL-17 enabled us to probe the impact of IL-23 and IL-17 inhibition alone and in combination and, for the first time, demonstrate a cooperative effect of dual inhibition on cytokine production in vitro and enhanced efficacy in vivo. These findings are remarkable given the established connection between IL-23 and IL-17 that might suggest that these cytokines are redundant (Murphy et al., 2003; Langrish et al., 2005; Luger et al., 2008). Yet, IL-23 induces additional cytokines, such as IL-22, that have distinct functions (Liang et al., 2006). Consistent with the biology of IL-23 in mice, we showed that committed Th17 cells stimulated with IL-23 secreted robust levels of IL-17A and IL-22. Notably, the secretion of IL-22 is unique to mouse Th17 cells polarized with TGF-β1 and IL-6. In human T cells, the addition of TGF-β has been shown to inhibit IL-22 but drive production of IL-26, which is related to IL-22 and is also induced by IL-23 (Wilson et al., 2007; Manel et al., 2008). Both IL-22 and IL-26 have been associated with human diseases, especially in psoriasis, in which both cytokines are elevated in lesional psoriatic skin (Wilson et al., 2007). In our studies, the production of IL-22 coincident with IL-17A in the mouse Th17 cells afforded two readouts that could be used to measure IL-23 function. Incubation with an unformatted anti–IL-23p19-selective adnectin, ATI-1221, as well as a PEGylated form, ATI-1249, potently and comparably inhibited both IL-23–dependent cytokines, IL-17A and IL-22. In vivo, dosing ATI-1249 into IL-23–challenged mice also blocked production of IL-17 and IL-22. These data confirmed the in vitro and in vivo activity of these adnectins and the role for IL-23 in driving not only IL-17 but also IL-22.
In a similar way, IL-17 can act independently of IL-23. In mice, IL-17 can be produced in the absence of IL-23 by Th17 cells during differentiation, as well as by iNKT and mast cells (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006; Yoshiga et al., 2008; Hueber et al., 2010). To assess the impact of IL-17 blockade, two inhibitors of IL-17 were generated, a mouse decoy receptor, mIL-17RA-Fc, and a monoclonal antibody against murine IL-17A. Both agents blocked IL-17A/A– and IL-17A/F–induced responses in NIH/3T3 cells in vitro. Moreover, in an IL-17–dependent PD model in vivo, dosing the anti–IL-17 antibody into mice before challenge with IL-17A/A and IL-17A/F inhibited CXCL1 induction by either cytokine. Together, these data confirmed the activity of our key tool inhibitors for neutralizing IL-17A activity in vitro and in vivo and enabled us to assess the impact of any IL-23–independent functions of IL-17.
Using these inhibitors of IL-23 and IL-17, the relative effects of each agent alone and in combination was tested in a coculture system where exogenous IL-23 drove IL-17 production from Th17 cells, which in turn induced cytokine production from NIH/3T3 cells. Here, we chose to measure IL-6 rather than CXCL1 induction from NIH/3T3 cells on the basis of the demonstrated role for IL-6 in autoimmune disease (Yao et al., 1995; Hwang et al., 2004; Yen et al., 2006; Chiricozzi and Krueger, 2013). Using this system, we confirmed the key functional role of IL-23 for parallel induction of IL-22 and IL-17 and the key functional role of IL-17 in the induction of IL-6, but we also revealed some critical limitations of neutralizing either IL-23 or IL-17 alone. Blocking IL-23 only partially inhibited IL-17 produced by the cytokine and, therefore, had a modest nonsignificant effect on the IL-6 produced in the system. Conversely, inhibiting IL-17 was more efficacious for blocking IL-6 but did not inhibit IL-22 produced by IL-23. The combination treatment showed good neutralization of IL-22 and, interestingly, greater inhibition of IL-6 than either treatment alone, perhaps reflecting that when the level of IL-17 is decreased by IL-23 blockade, inhibition of the cytokine production by the residual IL-17 is more easily inhibited. Remarkably, even the basal IL-6 produced in the coculture system was extremely sensitive to IL-17 blockade, with a near-complete suppression of IL-6 by the anti–IL-17 antibody. The profound inhibition of proinflammatory cytokines with dual inhibition suggested that this comprehensive blockade might offer enhanced efficacy for treating disease.
To investigate whether dual inhibition of IL-17 and IL-23 might lead to improved efficacy in vivo, we tested the anti–IL-23 adnectin ATI-1249 and the anti–IL-17 antibody in the IMQ-induced skin inflammation model. Mice deficient in IL-23p19 or the IL-17 receptor are protected from IMQ-induced scaling and skin thickening, demonstrating a pivotal role for the IL-23/IL-17 axis in this model (van der Fits et al., 2009). Protection in IL-23p19–deficient mice stems from dramatically reduced IL-17A and IL-17F. In accord with these data, we showed that treatment with the anti–IL-23 adnectin in IMQ-challenged mice reduced skin thickening and scaling and, by histologic examination, scores for acanthosis and cell infiltrates. In contrast, treatment with the anti–IL-17 antibody had little or no effect on the IMQ-induced disease. The lack of efficacy was surprising given the demonstrated suppression of disease in IL-17 receptor–deficient mice. Yet, the selectivity of our antibody for IL-17A/A and IL-17A/F but not IL-17F/F could be responsible for this outcome (Supplemental Table 2) and are consistent with studies demonstrating an association of IL-17F with skin inflammation (van der Fits et al., 2009; Fujishima et al., 2010). Moreover, IL-17RA–deficient mice likely elicit a more comprehensive blockade of all forms of IL-17, including IL-17F, since IL-17A/A, IL-17A/F, and IL-17F/F all require IL-17RA for signaling (Gaffen, 2009). Follow-on studies using pharmacologic inhibitors of IL-17A and IL-17F would be required to confirm this hypothesis. Nevertheless, neutralizing both IL-23 and IL-17 caused a cooperative effect to reduce IMQ-induced pathology, especially in disease scores for scaling and histologic scores for cellular infiltrate. These findings suggest that, although blocking IL-23 alone might significantly inhibit cytokine secretion and downstream responses, the added inhibition of IL-17A leads to a more robust blockade of cytokine, including any from IL-23–independent sources. Notably, the serum IL-6 and IL-22 levels induced in IMQ-challenged animals in the presence of inhibitors of both IL-23 and IL-17 were reduced comparably or more than either inhibitor alone. These in vivo data are remarkably consistent with our in vitro data demonstrating a similar cooperative effect of these inhibitors for reducing IL-6 and IL-22 levels. Taken together, our data confirm the role of IL-23 and IL-17 in IMQ-induced skin inflammation and demonstrate that dual blockade of both IL-23 and IL-17 offers enhanced efficacy against disease.
The involvement of IL-23 and IL-17 in skin inflammation has now been well validated by clinical trials in psoriasis, where treatments with antibodies against either IL-23 or IL-17 demonstrated impressive efficacy in patients. In phase II studies, antibodies against IL-23p19, IL-17, and the IL-17RA receptor subunit, all caused >75% disease clearance [Psoriasis Area and Severity Index (PASI) 75] in 70–80% of patients. Almost half also reached PASI 100 or 100% disease clearance (Papp et al., 2012a,b, 2013a,b; Rich et al., 2013; Gaffen et al., 2014). Our data showing efficacy from targeting IL-23 alone in the IMQ-induced model substantiate the role of IL-23 in both mouse and human skin inflammation. Yet, the lack of efficacy from neutralizing IL-17 alone in the IMQ model highlight potential differences between the animal and human pathology. Antibodies against IL-17A specifically are highly efficacious for treating psoriasis, suggesting a significant role for IL-17A in human but not mouse disease (Papp et al., 2012a,b; Patel et al., 2013; Rich et al., 2013). It is probable that, although IL-23 and IL-17 levels are known to be elevated in the lesional skin of psoriasis patients, the relative involvement of each in the development of the disease in mice and humans is different (Nograles et al., 2008; Johansen et al., 2009; Martin et al., 2013). Because the mouse model does not precisely parallel human disease, the direct applicability of our findings to psoriasis remains uncertain. Nevertheless, the cell types, cytokines, and mechanisms involved in the IMQ model are comparable to those in human disease. Therefore, our data suggest that blocking IL-23 and IL-17 simultaneously might offer superior anti-inflammatory activity against some elements of skin inflammation. Still, the magnitude of the response to all the mechanisms targeting the IL-23/IL-17 pathway in clinical trials raises the question of whether there is room in human psoriasis for increased efficacy through a more comprehensive targeting of these pathways.
Beyond skin pathology, a critical role for both IL-23 and IL-17 has also been described in models of CNS inflammation, leading us to investigate whether dual targeting might offer an enhanced efficacy in the MOG peptide–induced EAE model. Mice that are genetically deficient for IL-23p19 or IL-17, or receive treatment with IL-23p19 or IL-17 neutralizing agents, have shown reduced disease in various EAE models (Hofstetter et al., 2005; Langrish et al., 2005; Chen et al., 2006; Kroenke et al., 2008; Ishigame et al., 2009; Hu et al., 2010; El-Behi et al., 2011; Kap et al., 2011). Consistent with these data, treatment of MOG peptide–challenged mice with either the anti–IL-23 adnectin alone or the anti–IL-17 antibody alone significantly abrogated disease compared with controls as measured by decreased clinical score, maintenance of body weight, and histology readouts of reduced inflammation and demyelination. Even though each of the inhibitors alone demonstrated a reduction in the disease, the combination treatment dramatically reduced all disease manifestations to levels significantly lower than the suppression by either ATI-1249 or the anti–IL-17 antibody alone. This outcome further supports our assertion that a dual blockade of IL-23 and IL-17 offers better efficacy than blocking either cytokine alone in murine models of CNS inflammation.
Though clinical trials testing neutralizing agents against either IL-23 or IL-17 yielded uniformly positive outcomes in psoriasis patients, the results from these inhibitors in MS patients have been mixed. Despite the universal effect of IL-23 or IL-17 neutralization in reducing EAE, only the IL-17 antibodies have shown early positive data in clinical trials of MS, and trials with agents against IL-12p40 failed to demonstrate any efficacy (Segal et al., 2008; Longbrake and Racke, 2009). These findings may be surprising, given the significant impact of both anti–IL-23p19 and anti–IL-17 antibodies for preventing CNS inflammation in mice, but they highlight again the discrepancies between the mouse models and human disease (Chen et al., 2006; Uyttenhove and Van Snick, 2006). Yet, IL-23 and IL-17 brain expression levels have been associated with MS, in which IL-23p19 is expressed in active MS lesions and IL-17A is overexpressed in patient brain biopsies (Li et al., 2007; Tesmer et al., 2008). As with the IMQ model, the cell types and cytokines involved in the EAE model are comparable to those in human disease. Therefore, our data demonstrating a remarkably suppressive effect of dual IL-23 and IL-17 neutralization of EAE disease may suggest that a similar combination could lead to more promising efficacy in MS relative to the modest or no efficacy seen clinically with the individual agents.
Collectively, our findings demonstrate that comprehensive neutralization of both IL-23– and IL-17–dependent cytokines through dual blockade of IL-23 and IL-17 leads to enhanced efficacy for reducing disease in the IMQ skin inflammation and MOG peptide–induced EAE disease models. Comparison of our data with clinical data in psoriasis and MS for anti–IL-23 and anti–IL-17 therapies, as well as the published data in murine models of skin and CNS inflammation, suggests that the balance of the role of IL-17 and IL-23 may be different in animal models compared with human disease. However, assessment of the dual targeting of IL-17 and IL-23 in these murine models does provide proof of principle and suggests that, even given the potential human-to-murine difference, the more complete blockade obtained with dual targeting will provide better efficacy in human disease. These data provide a basis for pursuing a dual targeting strategy for IL-23 and IL-17, either through separate dosing of neutralizing agents or perhaps with a bispecific molecule. The generation of a stable bispecific molecule with specificity for IL-23 and IL-17 has been described and while the function of each IL-23 and IL-17 neutralizing arm was demonstrated in separate assays there was no validation that the combined effect of neutralizing both IL-17 and IL-23 was more efficacious than monotherapies either in vitro or in vivo (Mabry et al., 2010). Our data from both in vitro and in vivo models suggest that a more complete blockade of IL-23– and IL-17–dependent responses can be achieved through dual inhibition and that this profound inhibition of Th17 responses might offer enhanced benefit for treating certain autoimmune disorders.
Authorship Contributions
Participated in research design: Mangan, Su, Ditto, Lin, Yang, Cotter, Shuster, Borowski, Carvajal, Das Gupta, Xie, Zhao.
Conducted experiments: Mangan, Jenny, Picarillo, Skala, Ditto, Yang, Cotter, Shuster, Borowski, Thomas.
Contributed new reagents or analytic tools: Su, Jenny, Tatum, Lin, Devaux, Das Gupta.
Performed data analysis: Mangan, Yang, Cotter, Shuster, Song, Borowski, McIntyre, Xie, Heimrich.
Wrote or contributed to the writing of the manuscript: Mangan, Su, Cotter, Shuster, Heimrich, Devaux, Carvajal, McIntyre, Xie, Struthers, Salter-Cid.
Footnotes
- Received March 6, 2015.
- Accepted May 22, 2015.
This study was supported by Bristol-Myers Squibb.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ANOVA
- analysis of variance
- BMS
- Bristol-Myers Squibb
- CNS
- central nervous system
- EAE
- experimental autoimmune encephalomyelitis
- ELISA
- enzyme-linked immunosorbent assay
- GM-CSF
- granulocyte monocyte colony-stimulating factor
- HAT
- hypoxanthine-aminopterin-thymidine
- IFN-γ
- interferon-γ
- IL
- interleukin
- IMQ
- imiquimod
- iNKT
- invariant natural killer T
- MOG
- myelin oligodendrocyte glycoprotein
- MS
- multiple sclerosis
- PBS
- phosphate-buffered saline
- PBST
- 1× PBS solution with Tween
- PD
- pharmacodynamic
- rm
- recombinant murine
- TGF
- transforming growth factor
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}