


Abstract
Administration of a high-dose regimen of methamphetamine (METH) rapidly and profoundly decreases plasmalemmal and vesicular dopamine (DA) transport in the striatum, as assessed in synaptosomes and purified vesicles, respectively. To determine whether these responses were common to other amphetamines of abuse, effects of methylenedioxymethamphetamine (MDMA) on the plasmalemmal DA transporter (DAT) and vesicular monoamine transporter-2 (VMAT-2) were assessed. Similar to effects of METH reported previously, multiple high-dose MDMA administrations rapidly (within 1 h) decreased plasmalemmal DA uptake, as assessed ex vivo in synaptosomes prepared from treated rats. Unlike effects of multiple METH injections, this deficit was reversed completely 24 h after drug treatment. Also in contrast to effects of multiple METH injections, 1) MDMA caused little or no decrease in binding of the DAT ligand WIN35428, and 2) neither prevention of hyperthermia nor prior depletion of DA prevented the MDMA-induced reduction in plasmalemmal DA transport. However, a role for phosphorylation was suggested because pretreatment with protein kinase C inhibitors attenuated the deficit caused by MDMA in an in vitro model system. In addition to affecting DAT function, MDMA rapidly decreased vesicular DA transport as assessed in striatal vesicles prepared from treated rats. Unlike effects of multiple METH injections reported previously, this decrease partially recovered by 24 h after drug treatment. Taken together, these results reveal several differences between effects of MDMA and previously reported METH on DAT and VMAT-2; differences that may underlie the dissimilar neurotoxic profile of these agents.
Methylenedioxymethamphetamine (MDMA; “Ecstasy”) has received considerable recent attention due to both its recreational use and neurotoxic potential. Its abuse has increased dramatically over the past several years. For instance, the percentage of 8th-graders reporting having used MDMA in the previous year increased from 1.7% in 1999 to 3.1% in 2000. Among high school seniors, usage increased from 5.6 to 8.2% (Johnston et al., 2000). Hence, an understanding of the consequences associated with administration of this stimulant/hallucinogen is important.
Many investigators have shown that high-dose administrations of amphetamine analogs, including MDMA, cause persistent changes in monoaminergic neuronal function, but with varying expressions. For example, multiple injections of methamphetamine (METH) cause dopamine (DA) deficits persisting weeks and months after drug treatment (Koda and Gibb, 1973; Seiden et al., 1976; Hotchkiss et al., 1979; Morgan and Gibb, 1980; Eisch et al., 1992). In contrast, MDMA is far less toxic to DA systems (Johnson et al., 1988; Insel et al., 1989). In addition, we demonstrated recently that multiple high-dose injections of each of these agents also cause a rapid (within 1 h) decrease in plasmalemmal DA transporter (DAT) function (Fleckenstein et al., 1997;Kokoshka et al., 1998; Metzger et al., 1998). We and others have demonstrated that high-dose METH administration rapidly decreases vesicular monoamine transport, as assessed in purified striatal vesicles prepared from treated rats (Brown et al., 2000, 2001; Hogan et al., 2000). The acute effects of METH are not due to residual drug introduced by the original drug treatment (Kokoshka et al., 1998). Effects of MDMA on vesicular transport have not been reported.
The DAT is a principal regulator of DA disposition (i.e., of intra- and extraneuronal DA concentrations), and changes in DA disposition resulting from amphetamine analogs putatively contribute to their ability to cause long-term DA deficits in the striatum. In particular, we (for review, see Fleckenstein et al., 2000) and others (Cubells et al., 1994; Fumagalli et al., 1999; LaVoie and Hastings, 1999) have hypothesized that psychostimulants may redistribute DA from the reducing environment within synaptic vesicles to extravesicular intracellular oxidizing environments, thus causing the formation of oxygen radicals and reactive metabolites within DA neurons that trigger selective DA terminal loss. Accordingly, DA-releasing agents that rapidly decrease DAT function (i.e., METH; Fleckenstein et al., 1997;Kokoshka et al., 1998) may interfere with DAT function and attenuate DA efflux, thereby “trapping” DA in intraneuronal spaces where it can damage DA nerve terminals. Hence, an understanding of the effect of psychostimulants on DAT is important.
In addition to DAT, VMAT-2 is a significant regulator of intraneuronal DA concentrations. Presumably, a decrease in the function of the VMAT-2 impedes the sequestration of DA into synaptic vesicles, and may, therefore, increase cytoplasmic DA concentrations. Accordingly, a stimulant-induced decrease in vesicular uptake would presumably contribute to effects leading to persistent DA neuronal deficits. Consistent with this hypothesis, increased METH neurotoxicity in heterozygous VMAT-2 knockout mice has been reported (Fumagalli et al., 1999). Hence, like with DAT, an understanding of the effects of stimulants on VMAT-2 is important.
Our laboratory has hypothesized that concurrent stimulant-induced decreases in plasmalemmal DAT and VMAT-2 function may contribute to persistent neurotoxic DA deficits by promoting intraneuronal DA accumulation and reactive oxygen species generation (for review, seeFleckenstein et al., 2000). Alterations in the ratio of DAT to VMAT-2 function have been implicated in other disease states as well, including the deficits associated with Parkinson's disease (for review, see Miller et al., 1999). Accordingly, differences in the effects of METH and MDMA on DAT and VMAT-2 function may contribute to their dissimilar long-term neurotoxic DA consequences. Hence, the purpose of the present study was to investigate the impact of MDMA on DAT and VMAT-2, and compare these data with effects of METH as reported previously (Kokoshka et al., 1998; Brown et al., 2000, 2001; Metzger et al., 2000). Because it has been demonstrated that activation of protein kinase C (PKC) can alter the impact of psychostimulants on DAT function (Kantor and Gnegy, 1998; Sandoval et al., 2001), and that activation of DA receptors effects METH-induced alterations in plasmalemmal (Metzger et al., 2000) and vesicular (Fleckenstein et al.; 2001) DA uptake, the role of PKC and DA receptors in effecting MDMA-induced changes in either plasmalemmal or vesicular DA uptake was assessed. Results reveal that MDMA administration rapidly and reversibly decreases both plasmalemmal and vesicular DA transport. Mechanisms underlying these phenomena per se and in comparison with previously reported effects of METH are discussed.
Experimental Procedures
Materials.
3,4-MDMA hydrochloride and (−)-cocaine hydrochloride were generously supplied by the National Institute on Drug Abuse (Bethesda, MD). (−)-Eticlopride hydrochloride, pargyline hydrochloride, α-methyl-p-tyrosine hydrochloride (αMT), and (S-2,6-diamino-N-[[1-oxotridecyl)-2-piperidinyl]methyl]-hexanamide dihydrochloride (NPC15347) were purchased from Sigma Chemical (St. Louis, MO). 2-[1-3(Aminopropyl)indol-3-yl]-3(1-methylindol-3-yl)maleimide, acetate (Ro31-7549) was purchased from Calbiochem (San Diego, CA). [7,8-3H]DA (49 Ci/mmol) was purchased from Amersham Biosciences, Inc. (Piscataway, NJ). [N-methyl-3H]WIN35428 (84.5 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). α-[2-3H]Dihydrotetrabenazine ([3H]DHTBZ; 20 Ci/mmol) was purchased from American Radiolabeled Chemicals (St. Louis, MO). Tetrabenazine was kindly donated by Drs. Jeffrey Erickson, Helene Varoqui (Louisiana State University Health Sciences Center, New Orleans, LA), and Erik Floor (University of Kansas, Lawrence, KS).
Animals.
Male Sprague-Dawley rats (270–350 g; Simonsen Labs, Gilroy, CA) were maintained under controlled light and temperature conditions, with food and water provided ad libitum. On the day of the experiment, rats were housed in groups (usually eight per cage) in plastic cages and were maintained in an ambient temperature of 24°C. Sixteen rats were used in each treatment group. Where indicated in figure legends, some cages were placed in a cool environment (ambient temperature 6°C) upon treatment with MDMA or saline to manipulate body temperature (i.e., to prevent the hyperthermia caused by MDMA treatment). Core (rectal) body temperatures were recorded using a digital thermometer (Physiotemp Instruments, Clifton, NJ) in all experiments in which ambient temperature was manipulated. For experiments in which rats received multiple administrations of MDMA, rectal temperatures were recorded immediately before the first MDMA or saline administration (t = 0 h) and every hour thereafter (t = 0–7 h). Drugs were administered as indicated in the legends of the appropriate figures, and doses were calculated as the respective free bases. All procedures were conducted in accordance with National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and the University of Utah Institutional Animal Care and Use Committee.
[3H]DA Uptake via Plasmalemmal Transporters and [3H]WIN35428 Binding.
Uptake of [3H]DA was determined in striatal synaptosomes prepared according to the method described by Kokoshka et al. (1998). Briefly, fresh striatal tissue was homogenized in cold 0.32 M sucrose and centrifuged (800g for 12 min; 4°C). The supernatant (S1) was then centrifuged (22,000g for 15 min; 4°C), and the resulting pellet (P2) was resuspended in ice-cold modified Krebs' buffer (126 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 16 mM sodium phosphate, 1.4 mM MgSO4, 11 mM dextrose, 1 mM ascorbic acid, pH 7.4). Assays were conducted in Krebs' buffer. Each assay tube contained synaptosomal tissue (i.e., resuspended P2 obtained from 1.5 mg of original wet weight striatal tissue) and 1 μM pargyline. Nonspecific values were determined in the presence of 100 μM cocaine. After preincubation of assay tubes for 10 min at 37°C, assays were initiated by the addition of [3H]DA (0.5 nM final concentration). Samples were incubated at 37°C for 3 min. Samples were then filtered through Whatman GF/B filters (Brandel Inc., Gaithersburg, MD) soaked previously in 0.05% polyethylenimine. Filters were washed rapidly three times with 3 ml of ice-cold 0.32 M sucrose by using a Brandel filtering manifold. Radioactivity trapped in filters was counted using a liquid scintillation counter. Remaining resuspended P2 samples were assayed for protein concentrations according to the method of Lowry et al. (1951). In MDMA in vitro experiments, samples were preincubated with 10 μM MDMA for 30 min at 37°C. After 30 min, resuspended P2 fractions were “washed” by centrifugation (22,000g for 15 min; 4°C). The resulting pellet (P3) was then resuspeded in ice-cold Krebs' buffer and once again centrifuged (22,000g for 15 min; 4°C) to obtain a P4 pellet that was subsequently resuspended and assayed. [3H]WIN35428 binding (0.5 nM final concentration) was conducted in phosphate-buffered 0.32 M sucrose, pH 7.4, with synaptosomes obtained from 2 mg (original wet weight) of striatal tissue per reaction tube, and samples were incubated on ice for 2 h. Samples were then filtered through Whatman GF/B filters (Brandel Inc.) soaked previously in 0.05% polyethylenimine. Filters were washed rapidly three times with 3 ml of ice-cold 0.32 M sucrose by using a Brandel filtering manifold. Radioactivity trapped in filters was counted using a liquid scintillation counter. Remaining resuspended P2 samples were assayed for protein concentrations according to the method of Lowry et al. (1951).
[3H]DA Uptake via Vesicular Monoamine Transporters and [3H]DHTBZ Binding.
Synaptic vesicles were obtained from synaptosomes prepared from rat striatum as described above. Synaptosomes were resuspended and homogenized in cold distilled deionized water. Osmolarity was restored by addition of 245 mM HEPES and 100 mM potassium tartrate (final concentrations; pH 7.5). Samples were centrifuged for 20 min at 20,000g (4°C) to remove lysed synaptosomal membranes. MgSO4 (1 mM, final concentration) was added to the supernatant, which was then centrifuged for 45 min at 100,000g (4°C). The resulting vesicular pellet was resuspended in wash buffer at a concentration of 50 mg/ml (original tissue weight). Based on published reports with similar protocols for vesicle preparation (Kadota and Kadota, 1973;Teng et al., 1997), we believe vesicles isolated in these studies to be of the small synaptic vesicle size (∼50 nM), the predominant type found in dopaminergic terminals in the striatum (Nirenberg et al., 1997). Vesicular [3H]DA uptake was performed by incubating 100 μl of synaptic vesicle samples (∼2.5 μg of protein) at 30°C for 3 min in assay buffer (final concentration: 25 mM HEPES, 100 mM potassium tartrate, 1.7 mM ascorbic acid, 0.05 mM EGTA, 0.1 mM EDTA, 2 mM ATP-Mg2+, pH 7.5) in the presence of [3H]DA (30 nM final concentration). The reaction was terminated by addition of 1 ml of cold wash buffer (assay buffer containing 2 mM MgSO4 substituted for the ATP-Mg2+, pH 7.5) and rapid filtration through Whatman GF/F filters soaked previously in 0.5% polyethylenimine. Filters were washed three times with cold wash buffer by using a Brandel filtering manifold. Radioactivity trapped in filters was counted using a liquid scintillation counter. Nonspecific values were determined by measuring vesicular [3H]DA uptake at 4°C in wash buffer. Binding of [3H]DHTBZ was performed as described by Teng et al. (1998). Briefly, 200 μl of the synaptic vesicle preparation (∼6 μg of protein) was incubated in wash buffer in the presence of [3H]DHTBZ (2 nM final concentration) for 10 min at 25°C. The reaction was terminated by addition of 1 ml of cold wash buffer and rapid filtration through Whatman GF/F filters soaked in 0.5% polyethylenimine. Filters were washed three times with ice-cold wash buffer. Nonspecific binding was determined by coincubation with 20 μM tetrabenazine. All protein concentrations were determined by a Bio-Rad protein assay (Bio-Rad, Hercules, CA).
DA Content.
On the day of the assay, frozen tissue samples were thawed, sonicated for 3 to 5 s in tissue buffer [0.05 M sodium phosphate/0.03 M citric acid buffer with 15% methanol (v/v); pH 2.5], and centrifuged for 15 min at 22,000g. Tissue pellets were retained and protein determined according to the method of Lowry et al. (1951). The supernatant was centrifuged a second time for 10 min at 22,000g. Supernatant (20 μl) was injected onto a high-performance liquid chromatograph system coupled to an electrochemical detector (+0.73 V) for separation and quantitation of dopamine levels by using the method of Chapin et al. (1986).
Statistics.
Statistical analyses were conducted in studies involving two groups by using a Student's t test. In studies involving three or more groups, analyses of variance were performed followed by a Fisher's protected least-significant difference post hoc comparison. Differences were considered significant if probability of error was p < 0.05.
Results
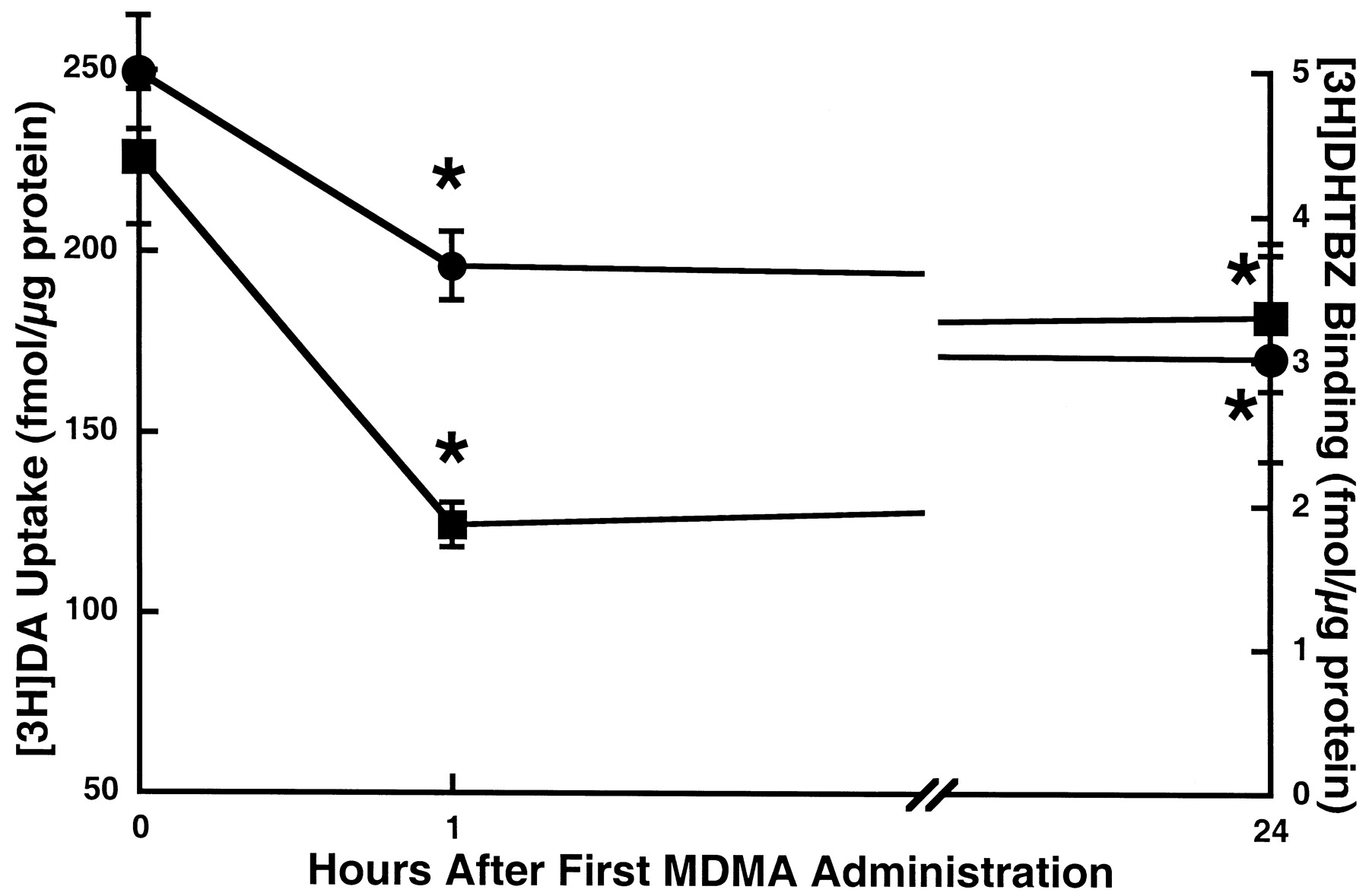
Results presented in Fig. 1 confirm previous reports that multiple high-dose administrations of MDMA rapidly (within 1 h) decrease plasmalemmal DA uptake function, as assessed in synaptosomes prepared from treated rats compared with synaptosomes prepared from saline vehicle-treated controls. This deficit represents a decrease in Vmax(2388 and 1410 fmol/mg of original wet weight tissue per 5 min for saline- and MDMA-treated rats, respectively), whereas transporterKm was virtually unaffected (99.6 versus 98.9 nM for saline- and MDMA-treated rats, respectively; Metzger et al., 1998). To determine the duration and nature of this phenomenon, the time-response effects of MDMA on plasmalemmal DA uptake and binding of the DAT ligand WIN35428 were conducted. Results reveal that the MDMA-induced deficit in plasmalemmal DA uptake was reversed 24 h after drug treatment. In contrast, WIN35428 binding was only slightly reduced (i.e., by 10%) 1 h after treatment: this deficit persisted 24 h after drug treatment.

Time-response effect of multiple MDMA administrations on striatal plasmalemmal [3H]DA uptake (▪) and [3H]WIN35428 binding (●). Rats received four injections (2-h intervals) of MDMA (10 mg/kg/injection s.c.) or saline vehicle (1 ml/kg/injection s.c.; zero time controls). MDMA-treated rats were decapitated 1 or 24 h, and saline-treated rats 1 h, after the final injection. Symbols represent the means and vertical lines 1 S.E.M. of determinations in six to eight rats. ★, values for MDMA-treated rats that are significantly different from saline-treated controls (p < 0.05).
Multiple administrations of MDMA (4 × 10 mg/kg; 2-h intervals; s.c.) to rats typically increases core body temperature by approximately 2–4°C. Previous studies have demonstrated that such hyperthermia contributes to the deficit in plasmalemmal DA uptake caused by multiple administrations of METH (Metzger et al., 2000). Hence, the role of body temperature in the reduction in plasmalemmal DA uptake induced by multiple administrations of MDMA was assessed by preventing the MDMA-induced increase in body temperature. Upon administration of MDMA, some rats were exposed to an ambient temperature of 6°C for the duration of the experiment (to maintain normothermic body temperature), whereas other MDMA-treated rats remained exposed to room temperature (24°C) to allow hyperthermia to occur. As shown in Fig. 2A, attenuation of MDMA-induced hyperthermia did not prevent the rapid decrease in [3H]DA uptake induced by multiple administrations of MDMA, compared with saline vehicle-treated controls. In this experiment, WIN35428 binding was not affected by either MDMA administration or by manipulating body temperatures (data not shown). Corresponding rat core body temperatures are shown in Fig. 2B.

A, effect of core body temperature on the decrease in striatal plasmalemmal [3H]DA uptake caused by multiple administrations of MDMA. Rats were maintained in an ambient temperature of 24°C before treatment. Upon receiving MDMA (4 × 10 mg/kg s.c.; 2-h intervals) or saline (1 ml/kg s.c.; 2-h intervals), rats were exposed to 6 or 24°C ambient temperature for the duration of the experiment. Rats were decapitated 1 h after the last MDMA or saline administration. B, time course of core body temperatures. Downward arrows represent time points of MDMA or saline administration. ★, values for MDMA-treated rats that are significantly different from saline-treated controls (p < 0.05).
In addition to demonstrating a role for hyperthermia, previous studies have shown that DA contributes to the deficit in DAT function caused by multiple administrations of METH (Metzger et al., 2000). Hence, the role of DA in the reduction of plasmalemmal DA uptake induced by multiple administrations of MDMA was assessed by depleting striatal DA levels by administering the tyrosine hydroxylase inhibitor αMT before MDMA treatment. αMT (150 mg/kg i.p.) was injected 5 and 1 h before, and 3 h after the first injection of MDMA. Striatal DA levels were greatly reduced by αMT pretreatment (55.0 ± 5.0 versus 10.1 ± 2.0 pg/μg protein for saline- versus αMT-treated rats, respectively; p < 0.05). As demonstrated in Fig.3A, pretreatment with αMT did not affect the MDMA-induced decrease in DAT activity. In this experiment, WIN35428 binding was decreased by 18% after MDMA treatment: αMT pretreatment did not prevent this deficit (Fig. 3B).

Effect of αMT on the decrease in striatal plasmalemmal [3H]DA uptake (A) and [3H]WIN35428 binding (B) caused by multiple administrations of MDMA. αMT (150 mg/kg i.p.) was administered 5 and 1 h before and 3 h after the first injection of MDMA. Rats received four injections (2-h intervals) of MDMA (10 mg/kg/injection s.c.) or saline vehicle (1 ml/kg/injection s.c.) and were decapitated 1 h later. Columns represent the means and vertical lines 1 S.E.M. of determinations in six to eight rats. ★, values for MDMA-treated rats that are significantly different from saline-treated controls (p < 0.05).
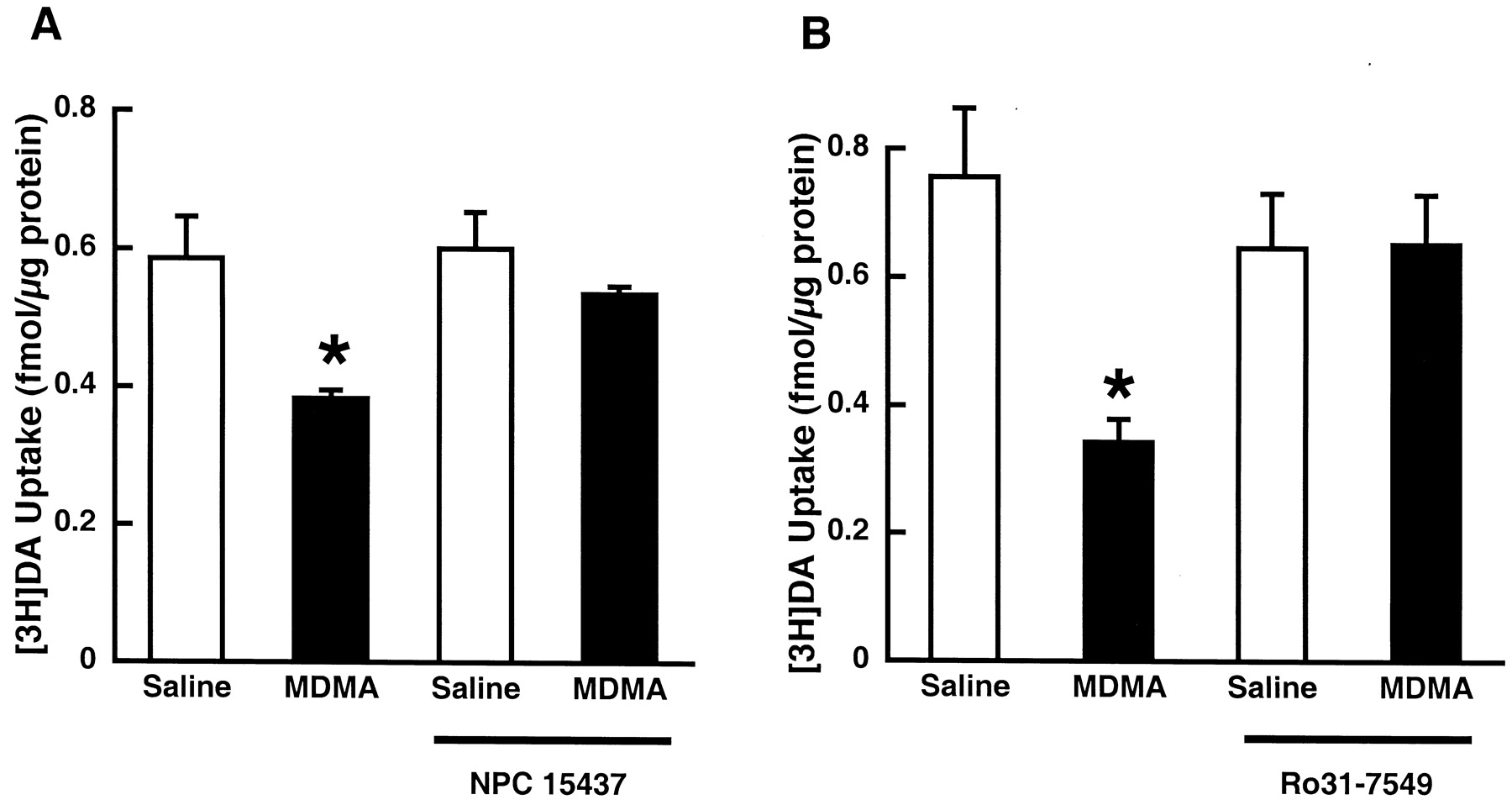
To elucidate the mechanism(s) whereby MDMA decreases DAT function in vitro, striatal synaptosomes were incubated with MDMA (10 μM) for 30 min at 37°C. A similar incubation paradigm demonstrated that this in vitro model appears to model some effects of METH treatment on DAT in vivo (Sandoval et al., 2001). Results presented in Fig.4 demonstrate that MDMA treatment also decreases DA uptake in vitro with a magnitude similar to that observed after multiple in vivo administrations of MDMA (i.e., 35–55%; compare with Figs. 1-3). Pretreatment with the PKC inhibitor NPC15437 attenuated the MDMA-induced deficit caused by in vitro incubation with MDMA (Fig. 4A). Moreover, pretreatment with another selective PKC inhibitor, Ro31-7549, attenuated the MDMA-induced deficit in vitro as well (Fig. 4B). Incubation of synaptosomes with MDMA had no effect on WIN35428 binding (data not shown).

Effects of NPC15437 (A) and Ro31-7549 (B) pretreatment on the decrease in plasmalemmal [3H]DA uptake in striatal synaptosomes induced by MDMA preincubation. Striatal synaptosomes were pretreated with 10 μM NPC 15437 or 10 μM Ro31-7549 for 5 min and subsequently exposed to 10 μM MDMA or assay buffer for 30 min at 37°C. ★, values for MDMA-treated preparations that are significantly different from saline-treated controls (p < 0.05).
To examine the effects of MDMA treatment on vesicular DA uptake, a time-response study was conducted. Results presented in Fig.5 demonstrate that not only does MDMA treatment rapidly diminish plasmalemmal DA uptake but also striatal vesicular DA uptake, compared with saline-treated controls. Specifically, multiple MDMA administrations rapidly decreased vesicular DA uptake, as assessed in vesicles purified from striata of treated animals. This deficit partially recovered 24 h after drug treatment. In addition, MDMA treatment reduced binding of the VMAT-2 ligand [3H]DHTBZ, both 1 and 24 h after treatment.
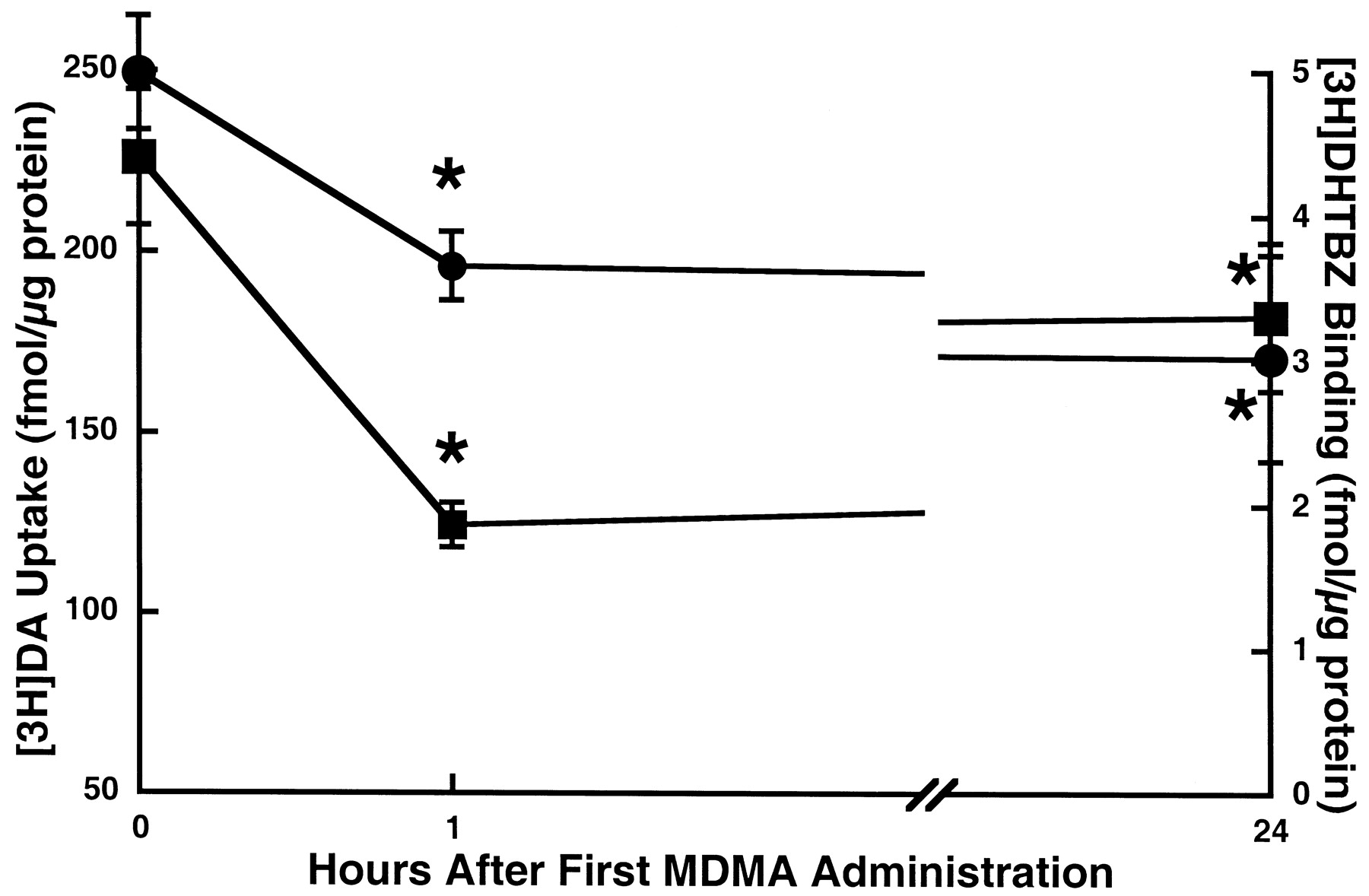
Time-response effect of multiple MDMA administrations on striatal vesicular [3H]DA uptake (▪) and [3H]DHTBZ binding (●). Rats received four injections (2-h intervals) of MDMA (10 mg/kg/injection s.c.) or saline vehicle (1 ml/kg/injection s.c.; zero time controls). MDMA-treated rats were decapitated 1 or 24 h, and saline-treated rats 1 h, after the final injection. Symbols represent the means and vertical lines 1 S.E.M. of determinations in six to eight rats. ★, values for MDMA-treated rats that are significantly different from saline-treated controls (p < 0.05).
Because it has been demonstrated that METH-induced hyperthermia contributes to the deficit in monoamine transporter function (Haughey et al., 2000; Metzger et al., 2000), the role of body temperature in the reduction in vesicular DA uptake induced by multiple administrations of MDMA was assessed by preventing the MDMA-induced increase in body temperature. Results presented in Fig.6, A and B, show that similar to the MDMA-induced effects on plasmalemmal DA transport, hyperthermia did not contribute to the drug-induced decrease in vesicular DA uptake or [3H]DHTBZ binding because its attenuation did not prevent these deficits. Corresponding rat core body temperatures for MDMA-treated rats, along with those of saline-treated controls, are shown in Fig. 6C, and are comparable to those reported previously after METH treatment (Metzger et al., 2000).

Effect of core body temperature on the decrease in striatal vesicular [3H]DA uptake (A) and [3H]DHTBZ binding (B) caused by multiple administrations of MDMA. Rats were maintained in an ambient temperature of 24°C before treatment. Upon receiving MDMA (4 × 10 mg/kg s.c.; 2-h intervals) or saline (1 ml/kg s.c.; 2-h intervals), rats were exposed to 6 or 24°C ambient temperature for the duration of the experiment. Rats were decapitated 1 h after the last MDMA or saline administration. C, time course of core body temperatures. Downward arrows represent time points of MDMA or saline administration. Columns/symbols represent the means and vertical lines 1 S.E.M. of determinations in six to eight rats. ★, values for MDMA-treated rats that are significantly different from saline-treated controls (p < 0.05). #, values for MDMA/24°C-treated rats that are significantly different from MDMA/6°C-treated rats (p < 0.05).
In the next experiment, the role of DA in the MDMA-induced decrease in vesicular DA uptake was assessed. Because depletion of DA resulting from αMT treatment increases vesicular DA uptake per se (Brown et al., 2001), the tyrosine hydroxylase inhibitor was not used in this experiment. Instead the role of D2receptors was determined using the D2 antagonist eticlopride. Administration of eticlopride (0.5 mg/kg i.p.) 15 min before each MDMA injection attenuated the MDMA-induced decrease in vesicular DA uptake (values for saline/saline-, saline/MDMA-, eticlopride/saline-, and eticlopride/MDMA-treated rats of 227 ± 7, 147 ± 11*, 234 ± 20, and 190 ± 13*# fmol/μg of protein, respectively; *p ≤ 0.05 versus saline/saline-treated rats;#p ≤ 0.05 versus saline/MDMA-treated rats).
Discussion
It is well established that high-dose administration of the amphetamine analog METH causes DA deficits persisting months and even years after drug treatment in rodents, nonhuman primates, and perhaps humans (Buening and Gibb, 1974; Seiden et al., 1976; Hotchkiss et al., 1979; Morgan and Gibb, 1980; Eisch et al., 1992; Wilson et al., 1996; Villemagne et al., 1998). In contrast, administration of the amphetamine analog MDMA is far less toxic to DA systems (Johnson et al., 1988; Insel et al., 1989). Assuming that the VMAT-2 and DAT are important to these differential phenomena (see Introduction), differential effects of METH and MDMA on these transporters would be expected. Hence, the purpose of this study was to characterize the acute effects of MDMA on DAT and VMAT-2, and compare these data with previous reports of the effects of METH on these transporters.
The results of this study indeed reveal significant differences between the effects of multiple administrations of METH and MDMA (both administered at doses of 10 mg/kg s.c., four injections at 2-h intervals) on plasmalemmal DA uptake. Specifically, the magnitude of the decrease caused by MDMA treatment (35–55%) is less than that observed 1 h after multiple METH administrations (≈70–80%;Kokoshka et al., 1998; Fleckenstein et al., 1999; Metzger et al., 2000). Moreover, the decrease observed 1 h after MDMA treatment recovers completely after 24 h (Fig. 1), whereas the decrease caused by METH only recovers to ≈60% of control values (Kokoshka et al., 1998). In addition, at least one component of the deficit in plasmalemmal DA uptake caused by METH treatment is associated with a decrease in WIN35428 binding (Kokoshka et al., 1998), whereas multiple MDMA injections had little or no acute effect on the binding of the plasmalemmal DAT ligand. Finally, neither depletion of DA nor prevention of hyperthermia attenuated the acute effects of MDMA on plasmalemmal DA uptake (Figs. 2 and 3). This is in contrast to METH in that both hyperthermia and DA contribute, in part, to the deficit in plasmalemmal DA uptake caused by multiple administrations of the stimulant (Metzger et al., 2000).
Not only are there significant differences between the effects of METH and MDMA on plasmalemmal DA uptake but alsovesicular DA uptake. For instance, MDMA causes deficits that are lesser in magnitude than those observed after METH treatment (25–30% as shown in Figs. 5-6 for MDMA versus ≈65% after METH treatment; Brown et al., 2000). Moreover, the effect of MDMA was substantially reversed 24 h after treatment, whereas the deficits in vesicular DA uptake caused by multiple injections with METH largely persist 24 h later (Brown et al., 2000). Interestingly, DA contributes to the deficits in vesicular DA uptake caused by multiple MDMA injections, as demonstrated by findings that pretreatment with the D2 antagonist eticlopride attenuates slightly the MDMA-induced deficit. (The role of DA in the effect of multiple METH treatments has not been determined.) Hyperthermia did not appear to contribute to the MDMA-induced deficit in vesicular DA uptake because its attenuation did not prevent this deficit, although it is noteworthy that in this experiment, hyperthermia was not prevented completely by placing the animal in a cool environment and hence a contribution of temperature to this phenomenon cannot be eliminated absolutely.
As described in the Introduction, we (for review, see Fleckenstein et al., 2000) and others (Cubells et al., 1994; Fumagalli et al., 1999;LaVoie and Hastings, 1999) have hypothesized that psychostimulants, such as METH, may cause DA terminal loss by redistributing DA from the reducing environment within synaptic vesicles to extravesicular intracellular oxidizing environments, thus resulting in formation of DA-associated oxygen radicals and reactive metabolites within neurons. Accordingly, DA-releasing agents that rapidly decrease DAT function (i.e., METH; Fleckenstein et al., 1997; Kokoshka et al., 1998) may interfere with DAT function and attenuate DA efflux (i.e., interfere with the ability of METH to cause DA release through the DAT), thereby “trapping” DA in intraneuronal spaces where it can damage nerve terminals. In addition, stimulant-induced decreases in vesicular uptake would presumably increase cytoplasmic DA concentrations and, thereby, contribute to persistent DA neuronal deficits. Accordingly, it was predictable that multiple administrations of METH, a regimen that causes persistent DA deficits (Haughey et al., 1999), causes deficits in plasmalemmal and vesicular DA uptake that were greater in magnitude and/or duration than those observed after multiple MDMA administrations (i.e., a regimen similar to that which does not cause persistent DA deficits; Johnson et al., 1988). In addition, our laboratory has demonstrated that there are several components to the acute decrease in plasmalemmal DA uptake induced by multiple METH injections, and that the DA- and hyperthermia-dependent components of this phenomenon are likely associated with the long-term DA deficits caused by the stimulant (Metzger et al., 2000). Interestingly, multiple MDMA injections did not effect such components (i.e., the acute deficit in plasmalemmal DA uptake caused by MDMA treatment was independent of DA and hyperthermia). Hence, these differences in the acute effects of multiple METH and MDMA injections on plasmalemmal and vesicular DA uptake may contribute to the dissimilar DA neurotoxic potential of these agents.
It is interesting to note that although there were several differences between effects of multiple METH and MDMA administrations, the acute effects of a single METH injection (15 mg/kg) largely resemble the acute effects of multiple MDMA treatments. Specifically, both phenomena are 1) similar in magnitude (Fleckenstein et al., 1999), 2) reversed 24 h after treatment (Fleckenstein et al., 1997; Figs. 1 and 2), and 3) occur independently of DA and of drug-induced hyperthermia (Metzger et al., 2000; Figs. 2 and 3). Hence, it might be predicted that like multiple MDMA administrations, a single METH injection would not cause long-term DA deficits. Accordingly, it has been demonstrated that a single 15 mg/kg METH injection does not effect long-term decreases in tyrosine hydoxylase activity, an indicator of the integrity of DA neuronal function (Kogan et al., 1976).
It has been demonstrated in an in vitro synaptosomal model that pretreatment with the selective protein kinase C inhibitor NPC15437 (10 μM, a concentration 30-fold less than the IC50 for inhibition of both protein kinase A and calcium/calmodulin-dependent protein kinase; Sullivan et al., 1992) attenuates the decrease in plasmalemmal DA uptake caused by METH administration (Sandoval et al., 2001). Results presented in Fig. 4demonstrate that preincubation with NPC15437, as well as another PKC inhibitor (Ro-31-7549), prevents the deficits induced by MDMA application as well. One caveat in interpreting these studies is that it remains to be determined whether the in vitro effects of MDMA are the same as those observed after multiple administrations in vivo. Still, these data indicate that similar mechanisms may underlie the effects of a single METH and multiple MDMA treatments. Because it has been suggested that the deficit in plasmalemmal DA uptake induced after amphetamine and/or METH treatment may be associated with PKC activation and/or subsequent internalization of the plasmalemmal DAT (Saunders et al., 2000; Sandoval et al., 2001), the present data suggest that the MDMA-induced effect on DAT may involve these processes as well.
In conclusion, multiple administrations of MDMA and METH differentially alter plasmalemmal and vesicular DA uptake. Specifically, multiple METH injections appear to cause an “extra” component that is hyperthermia- and DA-dependent, and makes its effects greater in magnitude and duration than the effects of multiple MDMA administrations, possibly resulting in METH-induced damage to DA neurons. MDMA and METH differentially alter vesicular DA uptake as well. In particular, the MDMA-induced decrease in vesicular DA uptake does not persist as long as the deficits induced by METH treatment. In addition, these studies support the hypothesis that differential effects of stimulants on VMAT-2 and DAT may underlie the differential neurotoxic properties of the stimulants. Whether the deficit in DAT function contributes to the deficit in VMAT-2 function, or vice versa, remains to be established. Further study regarding the contributions of these phenomena to the differentially long-term consequences of administering these stimulants is warranted.
Footnotes
-
This study was supported by National Institute on Drug Abuse Grants DA11389, DA 00869, and DA 04222.
- Abbreviations:
- MDMA
- methylenedioxymethamphetamine
- METH
- methamphetamine
- DA
- dopamine
- DAT
- dopamine transporter
- VMAT-2
- vesicular monoamine transporter-2
- PKC
- protein kinase C
- αMT
- α-methyl-p-tyrosine hydrochloride
- DHTBZ
- dihydrotetrabenazine
- Received September 20, 2001.
- Accepted December 5, 2001.
- The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}