


Abstract
The allosteric enhancer PD 81,723, a 2-amino-3-benzoylthiophene derivative, has been shown to potentiate agonist binding to A1 adenosine receptors (A1AdoRs) and to enhance the functional effects of adenosine and adenosine analogs. The objective of this study was to determine whether the apparent agonist-independent effect of PD 81,723 observed in CHO cells stably expressing the recombinant human A1AdoR was due to the potentiation of the action of endogenous adenosine, to the presence of constitutive receptor activity and/or to the binding of PD 81,723 to the agonist binding site of the A1AdoR. The allosteric enhancer PD 81,723, the A1AdoR agonist (R)-N6-(2-phenylisopropyl)adenosine and adenosine all significantly inhibited forskolin-stimulated cAMP accumulation in intact cells and increased [35S]-5′-(γ-thio)triphosphate binding to cell membranes. The effects of adenosine on cAMP formation and [35S]-5′-(γ-thio)triphosphate binding were attenuated by adenosine deaminase, but the effects of PD 81,723 were not. In the presence of ADA, the A1AdoR antagonist 8-cyclopentyl-1,3-dipropylxanthine increased forskolin-stimulated cAMP accumulation in cells expressing the recombinant human A1AdoR but not in nontransfected CHO cells. In binding experiments, the agonist (R)-N6-(2-phenylisopropyl)adenosine, but not PD 81,723, significantly displaced the specific binding of the A1AdoR agonist [3H]-N6-cyclohexyladenosine and the antagonist [3H]-8-cyclopentyl-1,3-dipropylxanthine. The results of this study demonstrate that in CHO cells stably expressing the recombinant human A1AdoR, the agonist-independent effect of PD 81,723 is not due to potentiation of the action of endogenous adenosine or mediated by the binding of the allosteric enhancer to the agonist binding site of the recombinant human A1AdoR. It is possible that these effects are due to potentiation of constitutive receptor activity by PD 81,723.
The 2-amino-3-benzoylthiophene derivative PD 81,723 has been shown to enhance A1AdoR-mediated responses of adenosine and adenosine analogs in guinea pig isolated perfused hearts (Amoah-Aprakuet al., 1993; Kollias-Baker et al., 1994a,1994b), rat isolated atria (Mudumbi et al., 1993), rat hippocampal brain slices (Janusz et al., 1991) and cultured FRTL-5 rat thyroid cells (Bruns and Fergus, 1990). It has been hypothesized that PD 81,723 potentiates A1AdoR-mediated actions by enhancing the binding of agonists to the receptor (Bruns and Fergus, 1990; Kollias-Baker et al., 1994a). PD 81,723 has been shown to enhance the binding of adenosine agonists to A1AdoR but not A2AdoR in membranes from human, guinea pig, rat and dog brain tissue (Bruns and Fergus, 1990) and human and guinea pig cardiac tissue (Kollias-Baker et al., 1994a). These results are entirely consistent with those reported in functional studies (Bruns and Fergus, 1990; Kollias-Baker et al., 1994a). In the heart, where both A1AdoRs and A2AdoRs are present, PD 81,723 selectively enhances the A1AdoR- but not A2AdoR-mediated actions of adenosine agonists (Kollias-Baker et al., 1994a). In addition, in the heart (Kollias-Baker et al., 1994a) and in hippocampal brain slices (Janusz et al., 1991), PD 81,723 alone (i.e., in the absence of adenosine or A1AdoR agonists) does not mimic the effect of adenosine or A1AdoR agonists. Thus, in these preparations, PD 81,723 enhances agonist-mediated responses but by itself it has no effect.
In a recent study carried out in CHO cells stably expressing the recombinant human A1AdoR, PD 81,723 was found to increase (1) the fraction of receptors in the high-affinity binding state, (2) the concentration of GTPγS required to half maximally uncouple receptor/G proteins complexes, (3) the binding of an A1AdoR agonist and (4) the potency of that same agonist to decrease forskolin-stimulated cAMP accumulation (Bhattacharya and Linden, 1995). These findings are consistent with the results of previous studies in human and guinea pig cardiac membranes; rat, dog and guinea pig forebrain membranes; and cultured FRTL-5 cells (Bruns and Fergus, 1990;Kollias-Baker et al., 1994a). A provocative finding of the study in CHO cells was that PD 81,723 alone, in the absence of an agonist, inhibited forskolin-stimulated cAMP accumulation (Bhattacharya and Linden, 1995). This apparent agonist-independent effect of PD 81,723 was not present in functional studies in isolated guinea pig hearts (Amoah-Apraku et al., 1993; Kollias-Baker et al., 1994a, 1994b), rat isolated atria (Mudumbi et al., 1993) or rat hippocampal brain slices (Janusz et al., 1991) but was noted in cultured FRTL-5 cells (Bruns and Fergus, 1990). It was postulated that this effect of the allosteric enhancer was due to PD 81,723 potentiating the action of either endogenous adenosine in the media or constitutive receptor activity (Bhattacharya and Linden, 1995). Alternatively, this effect of the allosteric enhancer could have been mediated by the binding of PD 81,723 to the agonist binding site of the recombinant human A1AdoR. Which of these mechanisms, however, is responsible for this effect of PD 81,723 remains to be determined. Therefore, in the present study we sought to determine whether the apparent agonist-independent effect of PD 81,723 observed in CHO cells stably expressing the recombinant human A1AdoR was due to the potentiation of the action of endogenous adenosine, to the presence of constitutive receptor activity and/or to the binding of PD 81,723 to the agonist binding site of the A1AdoR.
Methods
Materials.
PD 81,723 was the generous gift of Dr. Noel Cusack (Discovery Therapeutics, Richmond, VA). The human A1AdoR cDNA (GenBank accession no. X68485) was cloned as a 1.4-kb HindIII/XbaI fragment into the expression vector pCMV5 (Mumby et al., 1990) and cotransfected with pWLneo (Stratagene, La Jolla, CA) into CHO-KI cells (American Type Culture Collection CCL 61, Rockville, MD) according to the calcium phosphate method. Neomycin-resistant colonies were selected in 1 mg/ml G-418 and screened for expression of A1 receptors using the A1AdoR agonist [3H]CHA binding. cAMP, GTPγS, GDP, Tris·HCl, hydroxyapatite, dithiothreitol, ADA and bovine adrenal extract were purchased from Sigma Chemical Co. (St. Louis, MO). CPX, CPA, and (R)-PIA were purchased from Research Biochemicals Inc. (Natick, MA). [3H]CHA, [3H]CPX, [3H]cAMP and [35S]GTP[S] were purchased from DuPont-New England Nuclear Research Products (Boston, MA). G-418, trypsin EDTA and HBSS were purchased from GIBCO Life Technologies (Gaithersburg, MD). Ham’s F-12 cell culture media and fetal bovine serum were obtained from Mediatech Inc. (Washington, D.C.).
Cell Culture
CHO cells stably expressing the recombinant human A1AdoR were grown as monolayers on 150-mm plastic culture dishes in Ham’s F-12 media supplemented with 10% fetal bovine serum in the presence of 0.5 mg/ml G-418 in an atmosphere of 5% CO2/95% air at 37°C. Cells were subcultured twice weekly after detachment using 0.25% trypsin and 1 mM EDTA in HBSS. Cells were used 1 day before confluency.
Protocols
Membrane preparations.
CHO cells stably expressing the recombinant human A1AdoR were collected from culture dishes, homogenized in 25 ml of ice-cold 50 mM Tris·HCl, pH 7.4, and centrifuged at 48,000 × g for 15 min. The membrane pellet was washed twice by resuspension in fresh buffer and centrifugation. Final pellets were resuspended in 50 mM Tris·HCl, pH 7.4.
Radioligand binding.
To determine the effect of the allosteric enhancer on the binding of ligands to the A1AdoR, CHO cell membranes (0.04–0.1 mg of protein) were incubated in a buffer solution (300 μl) containing 50 mM Tris·HCl and the radioligand ([3H]CHA or [3H]CPX) for 2 hr at 19°C to 22°C in the absence and presence of PD 81,723. Specific binding of the radioligand was determined by subtracting nonspecific binding from total binding. Nonspecific binding of [3H]CHA and [3H]CPX was determined using unlabeled CPA (10 μM) and CPX (10 μM), respectively. Experiments were carried out in buffer containing 2 units/ml ADA unless otherwise stated.
To determine the effect of the allosteric enhancer on [35S]GTP[S] binding to G proteins in the absence and presence of A1AdoR agonists, CHO cell membranes (0.05–0.1 mg of protein) were incubated for 45 min at 19°C to 22°C in buffer (200 μl) containing 50 mM Tris·HCl, pH 7.4, 1 mM EDTA, 1 mM MgCl2, 10 μM GDP, 1 mM dithiothreitol, 100 mM NaCl, 0.3 to 0.5 nM [35S]GTP[S] (∼50,000 cpm) and 0.5% bovine serum albumin in the presence and absence of PD 81,723 (Lorenzenet al., 1994). Experiments were carried out in buffer containing 2 units/ml ADA unless stated otherwise.
Incubations were terminated by the addition of 4 ml of ice-cold 50 mM Tris·HCl, pH 7.4, followed by collection of membranes onto Whatman GF/C glass-fiber filters by vacuum filtration. Filters were washed three times to remove unbound ligand. Filter disks containing trapped membrane protein and radioligand were placed in 4 ml of Scintiverse BD (Fisher Scientific, Norcross, GA), and the radioactivity was quantified using a liquid scintillation counter.
cAMP assays.
cAMP assays were carried out on cells in suspension or on attached cells plated in 12-well clusters. Results presented are from experiments performed with cells in suspension unless otherwise indicated.
For cell suspensions, cells were removed from culture plates by treatment with 1 mM EDTA in HBSS for 10 min. Cells were pelleted by centrifugation at 500 × g for 5 min, followed by resuspension in fresh HBSS and repeat pelleting. Cells were then resuspended in HBSS containing 2 units/ml ADA (unless stated otherwise). Drugs were added to microcentrifuge tubes containing aliquots of the cell suspension in a final volume of 300 μl. The tubes containing the drugs and the cells were then incubated for 6 min at 37°C. Incubations were terminated by transferring tubes to a boiling water bath for 5 min. Cellular debris was pelleted by centrifugation at 2000 × g for 5 min, and the supernates were transferred to new microcentrifuge tubes for determination of cAMP content. To determine the cellular content of cAMP, 50-μl aliquots of supernate were incubated in buffer (150 μl) containing 50 mM Tris·HCl, pH 7.4, [3H]cAMP (4 nM) and bovine adrenal cortex extract in a final volume of 200 μl for 1 hr at 0°C (Baker et al., 1985). Hydroxyapatite (75 μl; 50:50 v/v in water) was added to each tube, and the mixture was incubated for an additional 6 min at 0°C. The assay was terminated, and the radioactivity was quantified as described previously for radioligand binding assays.
For attached cells, culture medium was aspirated from cells grown in 12-well culture clusters as adherent monolayers, and warm (37°C) HBSS was added to each well in the cluster. After 6 min, the HBSS was aspirated, and fresh, warm HBSS containing appropriate drugs was added. This solution was aspirated at the end of 6 min of incubation and replaced immediately with 1 ml of ice-cold 50 mM HCl. cAMP contents of the acid extracts of cells were measured by radioimmunoassay. Cellular extract (100 μl), antibody to cAMP (the generous gift of Dr. Gary Brooker, Georgetown University), and 125I-labeled succinyl cAMP tyrosyl methyl ester (∼20,000 dpm) were mixed and incubated in glass tubes overnight at 2°C. Hydroxyapatite (75 μl; 50:50 v/v in water) was then added to each sample, and the mixtures were incubated for 10 to 30 min at 2°C. An assay was terminated by vacuum filtration of samples and collection of bound radioactivity on filter paper using a Brandel cell harvester. The radioactivity of antibody-bound125I-labeled cAMP ester absorbed to hydroxyapaite and trapped on filter paper was quantified with a gamma counter. cAMP content of sample extracts was estimated by comparison of sample results with results of parallel assays of standards of known cAMP content (0.06–16 nM).
Data Analysis
All values are expressed as mean ± S.E.M. unless otherwise stated. Statistical analysis of differences among values in experiments with multiple-comparison groups was based on analysis of variance followed by Bonferroni testing. For experiments with two comparison groups, statistical analysis was performed with a two-tailedt test (IN STAT MAC, GraphPAD Software, San Diego, CA). Differences between group mean values were considered significant at P ≤ .05. Binding parameters (i.e.,B max, K d, IC50, and K i) were determined using the radioligand binding analysis program LIGAND 4.0 (Elsevier-Biosoft).
Results
Effect of PD 81,723 on agonist and antagonist binding.
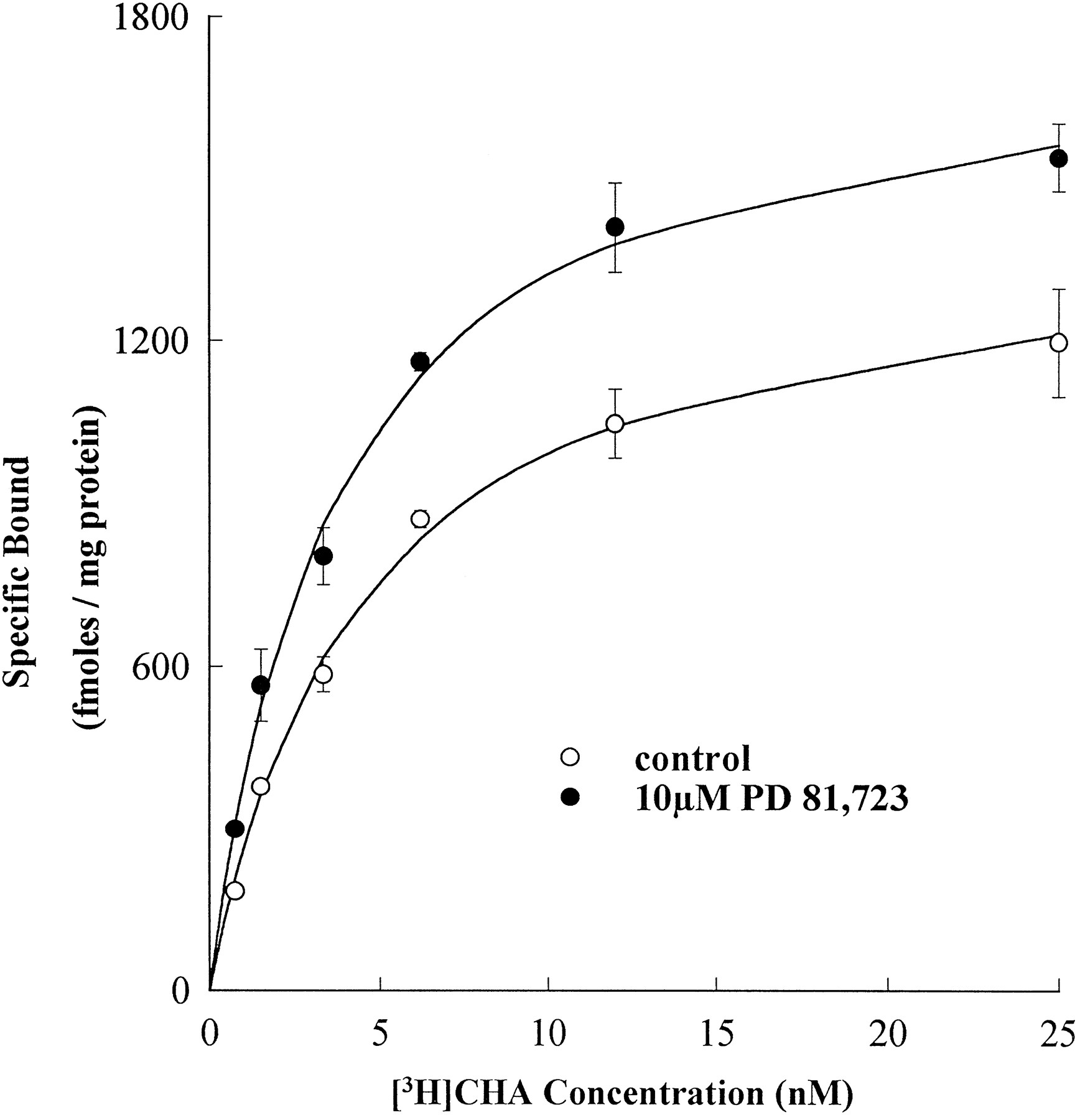
To determine whether the allosteric enhancer PD 81,723 potentiates agonist binding to A1AdoR, saturation binding experiments (n = 3) were performed using membranes prepared from CHO cells expressing recombinant human A1AdoR. As shown in figure 1, the maximum specific binding (B max) of the A1AdoR agonist [3H]CHA increased from 1306 ± 98 fmol/mg of protein in the absence to 1774 ± 105 fmol/mg of protein in the presence of PD 81,723 (10 μM; P ≤ .05). The concentration of [3H]CHA at which half of the receptors were occupied (K d) was not significantly (P ≥ .05) different in the absence (3.5 ± 0.3 nM) or presence (3.4 ± 0.2 nM) of PD 81,723 (10 μM). In contrast to the effect of PD 81,723 on agonist binding, the maximum specific binding (B max) of the A1AdoR antagonist [3H]CPX was not significantly (P ≥ .05) different in the absence (10,288 ± 240 fmol/mg of protein) and presence (10,474 ± 205 fmol/mg of protein) of PD 81,723 (10 μM;n = 3). In addition, the K d of [3H]CPX was also not significantly (P ≥ .05) different in the absence (1.7 ± 0.7 nM) or presence (1.4 ± 1.2 nM) of PD 81,723 (10 μM). The results of these experiments demonstrated that PD 81,723 significantly enhanced agonist, but not antagonist, binding to the recombinant human A1AdoR.

Effect of PD 81,723 on specific binding of the A1AdoR agonist [3H]CHA to membranes from CHO cells expressing recombinant human A1AdoR. The saturation isotherm revealed that PD 81,723 increased the maximum specific binding of [3H]CHA. Each data point represents mean ± S.E.M. specific binding of triplicate determinations from three experiments.
Affinity of the A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 for the recombinant human A1AdoR.
Competition binding experiments (n = 3) were carried out to determine the effects of (R)-PIA and PD 81,723 on the binding of the agonist radioligand [3H]CHA (2 nM) and the antagonist radioligand [3H]CPX (1 nM) to recombinant human A1AdoR. The A1AdoR agonist (R)-PIA decreased the specific binding of [3H]CHA (fig. 2A) and [3H]CPX (fig. 2B) in a concentration-dependent manner. The concentrations of (R)-PIA that displaced half of the specific binding (i.e., K i) of [3H]CHA and [3H]CPX were 7.7 ± 2.2 nM and 820 ± 104 nM, respectively (fig. 2). In contrast to the effect of the agonist (R)-PIA , the effect of the allosteric enhancer PD 81,723 on the specific binding of [3H]CHA was biphasic. The specific binding of [3H]CHA increased as the concentration of PD 81,723 was raised from 0.1 to 10 μM and fell when the concentration of PD 81,723 was further increased (fig. 2A). The allosteric enhancer did not, however, enhance the binding of the antagonist radioligand. PD 81,723, at concentrations >10 μM, decreased the specific binding of [3H]CPX (fig. 2B).

Effect of the A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 on specific binding of the agonist [3H]CHA (A) and the antagonist [3H]CPX (B) to membranes from CHO cells expressing recombinant human A1AdoR. Each data point represents mean ± S.E.M. specific binding of triplicate determinations from three experiments.
Effects of (R)-PIA and PD 81,723 on forskolin-stimulated cAMP accumulation.
Activation of the A1AdoR by adenosine agonists has been shown to decrease forskolin-stimulated cAMP accumulation in many different cell types (Altio et al., 1992; Ma and Green, 1992; Murphy et al., 1995). In CHO cells expressing the recombinant human A1AdoR, both the agonist (R)-PIA and the allosteric enhancer PD 81,723 inhibited forskolin-stimulated cAMP accumulation in a concentration-dependent manner in the presence of adenosine deaminase (ADA = 2 units/ml), an enzyme that catalyzes the degradation of adenosine to inosine (fig. 3). In a series of three experiments, forskolin (2 μM) increased cellular cAMP accumulation from a base-line level of 34 ± 12 pmol/mg to 823 ± 94 pmol/mg of cellular protein (fig. 3; control). The A1AdoR agonist (R)-PIA at concentrations of 0.5, 1 and 5 nM decreased forskolin (2 μM) stimulated cAMP accumulation to 46 ± 7%, 24 ± 6% and 14 ± 2%, respectively, of the control value (fig. 3; P ≤ .05). In a separate series of experiments (n = 3) in which forskolin (2 μM) increased cellular cAMP accumulation from a baseline level of 45 ± 9 pmol/mg to 797 ± 137 pmol/mg of cellular protein (control), PD 81,723 at concentrations of 5, 25 and 50 μM decreased forskolin (2 μM) stimulated cAMP accumulation to 49 ± 2%, 34 ± 3% and 24 ± 4%, respectively, of control (fig. 3; P ≤ .05).

Effect of the A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 on forskolin-stimulated cAMP accumulation in CHO cells expressing recombinant human A1AdoR. All studies were carried out in the presence of ADA (2 units/ml). Values are mean ± S.E.M. of three experiments. ∗Value is significantly less (P ≤ .05) than control (i.e., forskolin alone).
Effect of ADA on adenosine- and PD 81,723-mediated inhibition of forskolin-stimulated cAMP accumulation.
In CHO cells expressing the recombinant human A1AdoR, adenosine (0.5 μM) in the absence of ADA significantly (P ≤ .05) decreased forskolin (2 μM)-stimulated cAMP accumulation by 96 ± 3% (fig.4A; n = 3). In the same cells and in the absence of ADA, PD 81,723 (10 μM) significantly (P ≤ .05) decreased forskolin (5 μM)-stimulated cAMP accumulation by 50 ± 9% (fig. 4B; n = 3). In the presence of ADA (≥2 units/ml), however, adenosine failed to decrease forskolin-stimulated cAMP accumulation (fig. 4A). As shown in figure 4A, in the presence of adenosine (0.5 μM) and ADA at concentrations of 2, 5 and 10 units/ml, the cAMP accumulations caused by forskolin (2 μM) were 445 ± 50, 476 ± 52 and 496 ± 34 pmol/mg of cellular protein, respectively. These values were not significantly different (P ≥ .05) from the accumulation of cAMP in the presence of forskolin (2 μM) alone (538 ± 15 pmol/mg of cellular protein; fig. 4A). In contrast to the effect of adenosine, the inhibition of forskolin-stimulated cAMP accumulation caused by PD 81,723 was not attenuated by ADA (fig. 4B). The forskolin (5 μM)-stimulated cAMP accumulations in the presence of PD 81,723 (10 μM) alone and PD 81,723 (10 μM) and ADA (10 units/ml) were 429 ± 78 and 432 ± 69 pmol/mg of cellular protein, respectively. Both values were significantly (P ≤ .05) less than cAMP accumulation in the presence of forskolin (5 μM) alone (control; 854 ± 125 pmol/mg of cellular protein). Thus, ADA abolished the inhibitory effect of adenosine, but not PD 81,723, on forskolin-stimulated cAMP accumulation.

ADA attenuates adenosine-mediated, but not PD 81,723-mediated, inhibition of forskolin-stimulated cAMP accumulation in CHO cells expressing recombinant human A1AdoR. Values are mean ± S.E.M. of three experiments. ∗Value is significantly less (P ≤ .05) than control (i.e., forskolin alone).
Synergistic effect of (R)-PIA and PD 81,723 to inhibit forskolin-stimulated cAMP accumulation.
The A1AdoR agonist (R)-PIA (0.2 nM) and the allosteric enhancer PD 81,723 (2.5 μM) significantly (P ≤ .05) decreased forskolin (2 μM) stimulated cAMP accumulation from a control (i.e., in the absence of (R)-PIA and PD 81,723) value of 712 ± 26 pmol/mg to 543 ± 17 and 587 ± 14 pmol/mg of cellular protein, respectively (fig. 5; n = 3). In combination, however, the same concentrations of (R)-PIA and PD 81,723 decreased the forskolin (2 μM)-stimulated cAMP accumulation to 350 ± 12 pmol/mg of cellular protein. The decrease in cAMP caused by the combination of (R)-PIA and PD 81,723 was significantly greater (P ≤ .05) than the decrease caused by either agent alone and greater than the expected decrease caused by the additive effect of both agents together (fig. 5).

Synergistic effect of the A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 to decrease forskolin-stimulated cAMP accumulation in CHO cells expressing recombinant human A1AdoR. Forskolin (2 μM) increased cAMP from a base-line level of 54 ± 21 to 712 ± 26 pmol/mg of cellular protein (control). Both (R)-PIA (0.2 nM) and PD 81,723 (2.5 μM) alone significantly inhibited forskolin (2 μM)-stimulated cAMP accumulation. Together, (R)-PIA (0.2 nM) and PD 81,723 (2.5 μM) caused a significantly greater inhibition of cAMP accumulation. All studies were carried out in the presence of ADA (2 units/ml). Values are mean ± S.E.M. of determinations from three experiments. ∗ And ∗∗, values that are significantly different (P ≤ .05) from control (i.e., in the presence of forskolin alone) and (R)-PIA and PD alone, respectively.
Effect of CPX on forskolin-stimulated cAMP accumulation.
The effect of the A1AdoR antagonist CPX on cAMP accumulation was determined in nontransfected CHO cells and CHO cells expressing the recombinant human A1AdoR treated with 1 μM forskolin (n = 3). In the absence of CPX, cAMP accumulation in nontransfected cells treated with 1 μM forskolin (control) was 335 ± 15.0 nmol, which was significantly (P ≤ .05) greater than cAMP accumulation in CHO cells expressing A1AdoR (153 ± 17.0 nmol). As shown in figure 6, CPX increased cAMP accumulation in CHO cells expressing A1AdoR but not in nontransfected cells. In CHO cells expressing A1AdoR, CPX caused a concentration-dependent increase in cellular cAMP accumulation (fig. 6). CPX at concentrations of 3, 10 and 30 nM caused a 130 ± 4.4%, 175 ± 6.5% and 257 ± 15.5% increase in cAMP accumulation above control, respectively (fig.6). As shown in figure 6, CPX (3, 10 and 30 nM) had no effect on cAMP accumulation in nontransfected cells (P ≥ .05).

Effect of the A1AdoR antagonist CPX on forskolin-stimulated cAMP accumulations in nontransfected cells (CHO) and CHO cells expressing recombinant human A1AdoR (CHO/A1AdoR). Forskolin (1 μM) caused the accumulation of 335 ± 15 and 153 ± 17 nmol of cAMP in CHO and CHO/A1AdoR cells, respectively. In nontransfected cells (CHO), CPX had no effect on forskolin-stimulated cAMP accumulation. In contrast, in CHO/A1AdoR cells, CPX significantly increased forskolin-stimulated cAMP accumulations in a concentration-dependent manner. All studies were done in the presence of ADA (2 units/ml). Values are mean ± S.E.M. from three experiments. ∗Values are significantly greater (P ≤ .05) than control.
Effects of (R)-PIA and PD 81,723 on [35S]GTP[S] binding.
A1AdoR agonists have been shown to increase the specific binding of [35S]GTP[S] to Gi proteins in membranes prepared from bovine forebrain (Lorenzen et al., 1994). In membranes prepared from CHO cells expressing the recombinant human A1AdoR, both the A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 increased [35S]GTP[S] specific binding by almost 2-fold above base line in the presence of ADA (2 units/ml; n = 3). The increase in [35S]GTP[S] binding caused by the allosteric enhancer, however, was attenuated in the presence of the antagonist CPX, whereas the increase caused by the agonist was not (fig. 7). As shown in figure 7, in the absence of CPX, both (R)-PIA and PD 81,723 increased [35S]GTP[S] binding from 100% to ∼180%, almost a 2-fold increase. In the presence of CPX (100 nM), the baseline level of [35S]GTP[S] binding decreased from 100% to ∼60%. Nevertheless, (R)-PIA in the presence of CPX (100 nM) increased [35S]GTP[S] binding from 60% to 120%, a 2-fold increase (fig. 7A). In contrast to the agonist, PD 81,723 in the presence of CPX (100 nM) did not significantly (P ≥ .05) increase [35S]GTP[S] binding above control (fig. 7B).

Effect of the A1AdoR agonist (R)-PIA (A) and the allosteric enhancer PD 81,723 (B) on [35S]GTP[S] binding to membranes from CHO cells expressing recombinant human A1AdoR in the absence (○) and presence (•) of the antagonist CPX. All studies were done in the presence of ADA (2 units/ml). Each data point represents the mean ± S.E.M. specific binding of triplicate determinations from three experiments.
The A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 also acted synergistically to increase [35S]GTP[S] binding (fig. 8;n = 3). Neither (R)-PIA (1 nM) nor PD 81,723 (10 nM) alone significantly (P ≥ .05) increased [35S]GTP[S] binding (fig. 8). However, in combination, the same concentrations of (R)-PIA and PD 81,723 increased [35S]GTP[S] binding to 170 ± 6% of baseline (P ≤ .05; fig. 8).

Synergistic effect of the A1AdoR agonist (R)-PIA and the allosteric enhancer PD 81,723 to increase [35S]GTP[S] binding to membranes from CHO cells expressing recombinant human A1AdoR. Neither (R)-PIA (1 nM) nor PD 81,723 (10 nM) alone significantly increased [35S]GTP[S] binding above baseline. In combination, however, the same concentrations of (R)-PIA and PD 81,723 significantly (P ≤ .05) increased [35S]GTP[S] binding. All studies were done in the presence of ADA (2 units/ml). Values are mean ± S.E.M. of three experiments. ∗Value is significantly greater (P ≤ .05) than base line.
Effect of ADA on increases in [35S]GTP[S] binding caused by adenosine and PD 81,723.
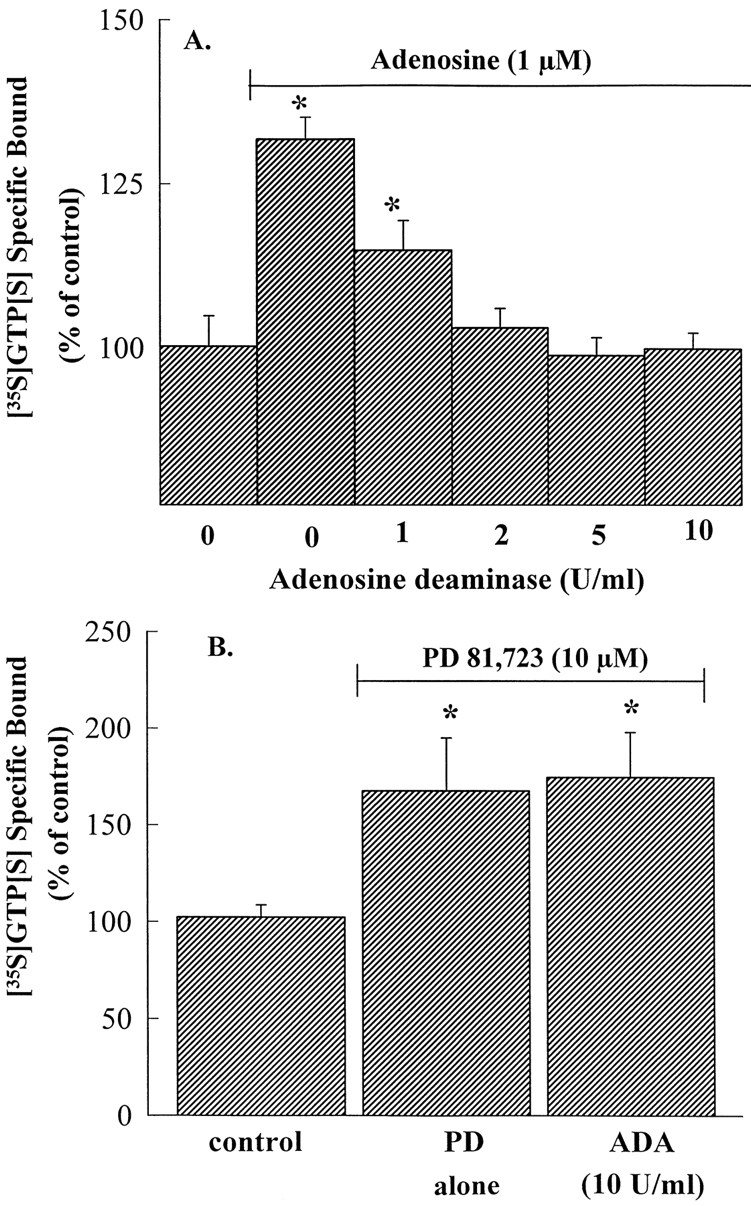
Adenosine (1 μM) and PD 81,723 (10 μM) increased [35S]GTP[S] binding (P ≤ .05) to CHO cell membranes expressing the recombinant human A1AdoR to 131 ± 7% and 167 ± 32% of baseline, respectively (fig.9; n = 3). However, as shown in figure9A, in the presence of ADA (≥2 units/ml), adenosine (1 μM) failed to increase [35S]GTP[S] binding above baseline (P ≥ .05). In contrast, the increase in [35S]GTP[S] binding caused by PD 81,723 was not attenuated by ADA (fig. 9B). As shown in figure 9B, [35S]GTP[S] binding was 167 ± 32% of baseline in the presence of PD 81,723 (10 μM) alone and 174 ± 27% of base line in the presence of PD 81,723 (10 μM) and ADA (10 units/ml); both values are significantly greater (P ≤ .05) than [35S]GTP[S] binding in the absence of PD 81,723 (i.e., control). Thus, ADA abolished the increase in [35S]GTP[S] binding caused by adenosine but not by PD 81,723.

Inhibition by ADA of the adenosine-mediated, but not PD 81,723-mediated, increase in [35S]GTP[S] binding to membranes from CHO cells expressing recombinant human A1AdoR. Adenosine (A) and PD 81,723 (B) increased [35S]GTP[S] binding. ADA (2–10 units/ml), in a concentration-dependent manner, reversed the adenosine-mediated increase in [35S]GTP[S] binding (A) but not the increase mediated by PD 81,723 (B). Values are mean ± S.E.M. of three experiments. ∗Values are significantly greater (P ≤ .05) than base line.
Discussion
The allosteric enhancer PD 81,723 has been shown to increase agonist- but not antagonist-specific binding to A1AdoR in membranes prepared from various tissues (i.e., heart and brain) from several different species (i.e., human, guinea pig, rat and dog), including recombinant human A1AdoRs (Bhattacharya and Linden, 1995; Bruns and Fergus, 1990; Kollias-Bakeret al., 1994a). Consistent with these findings, in the current study PD 81,723 increased the maximum specific binding (B max) of the agonist [3H]CHA to recombinant human A1AdoR in CHO cell membranes (figs. 1 and2). Also consistent with the results of previous studies, theB max value, as determined with the antagonist ligand [3H]CPX, was >9-fold higher than the value as determined with the agonist ligand [3H]CHA in the absence of PD 81,723 (Kollias-Baker et al., 1994a). This difference reflects the fact that the antagonist [3H]CPX labels the total population of A1AdoR, whereas the agonist [3H]CHA preferentially labels only those receptors in the agonist high-affinity state.
Similar to the results of radioligand binding studies, the potentiation of agonist-mediated A1AdoR responses by PD 81,723 is well described. For example, PD 81,723 has been shown to potentiate the effect of A1AdoR agonists in isolated guinea pig hearts (Amoah-Apraku et al., 1993; Kollias-Baker et al., 1994a, 1994b), rat isolated atria (Mudumbi et al., 1993) and rat hippocampal brain slices (Janusz et al., 1991). Consistent with these previous findings, in the current study in CHO cells stably expressing the recombinant human A1AdoR, PD 81,723 acted in synergism with the agonist (R)-PIA to inhibit forskolin-stimulated cAMP formation (fig. 5) and to increase [35S]GTP[S] binding (fig. 8). Inhibition of forskolin-stimulated cAMP formation is a measure of A1AdoR-mediated Gi protein activation (Cooperet al., 1980), whereas stimulation of [35S]GTP[S] binding has been shown to reflect GDP/GTP exchange that leads to Gi protein activation (Gilman, 1987;Lorenzen et al., 1994).
An important finding of this study was that PD 81,723 alone, in the absence of an agonist and in a concentration-dependent manner, inhibited forskolin-stimulated cAMP formation (fig. 3) and increased [35S]GTP[S] binding (fig. 7). These apparent agonist-independent effects of PD 81,723 have not been reported in preparations from studies evaluating the actions of this allosteric enhancer on A1AdoR expressed in native tissues, such as the isolated guinea pig heart and rat hippocampal brain tissue (Januszet al., 1991; Kollias-Baker et al., 1994b). These results are consistent, however, with those of two previous studies in which PD 81,723 appeared to inhibit forskolin-stimulated cAMP accumulation in cultured FRTL-5 cells (Bruns and Fergus, 1990) and in CHO cells stably expressing the recombinant human A1AdoR (Bhattacharya and Linden, 1995). The increase in [35S]GTP[S] binding mediated by PD 81,723 has not been previously reported.
There are several possible explanations for these apparent agonist-independent effects of PD 81,723. First, it is possible that the allosteric enhancer mediates these effects by binding to the agonist binding site of A1AdoR. The results of competition binding studies, however, demonstrated that none of the concentrations of PD 81,723 (1 pM to 100 μM) used in these studies significantly inhibited agonist binding to recombinant human A1AdoR. The specific binding of the agonist [3H]CHA increased as the concentration of PD 81,723 was raised from 0.1 to 10 μM and then fell when the concentration of PD 81,723 was increased further (fig. 2A). Unlike the effect of PD 81,723 on agonist binding, the allosteric enhancer did not increase the binding of the antagonist, and at a concentration of >50 μM, the specific binding of [3H]CPX decreased (fig. 2B). These results are consistent with previous reports indicating that the effect of PD 81,723 on agonist binding is biphasic, increasing agonist binding at lower concentrations but decreasing binding at higher concentrations (Bruns and Fergus, 1990). In contrast to the allosteric enhancer PD 81,723, the A1AdoR agonist (R)-PIA decreased the specific binding of both [3H]CHA and [3H]CPX in a concentration-dependent manner withK i values of 7.7 ± 2.2 and 820 ± 104 nM, respectively. The higher potency of the agonist (R)-PIA to compete with [3H]CHA, as opposed to [3H]CPX, for binding to A1AdoR in CHO cell membranes is due to the preferential binding of agonist to the high-affinity state of the receptor. Altogether, these results strongly support the conclusion that in CHO cells stably expressing the recombinant human A1AdoR, the effects of PD 81,723 alone (i.e., in the absence of an agonist) at concentrations of <100 μM are not due to the binding of PD 81,723 to agonist binding sites on A1AdoR.
A second possible explanation for the apparent agonist-independent effects of PD 81,723 is that the allosteric enhancer potentiates the action of endogenous adenosine present in the media. Therefore, to investigate this possibility, we sought to eliminate or reduce these effects of PD 81,723 using ADA to degrade adenosine present in the incubation media. In the presence of ADA, adenosine-mediated inhibition of cAMP accumulation (fig. 4A) and stimulation of [35S]GTP[S] binding (fig. 9A) were attenuated. In contrast, neither the inhibition of cAMP accumulation (fig. 4B) nor the stimulation of [35S]GTP[S] binding (fig. 9B) caused by PD 81,723 was attenuated by ADA at a concentration 5-fold higher than that required to attenuate the actions of the exogenous adenosine. Therefore, these results indicate that if the agonist-independent effect of PD 81,723 was due to potentiation of the action of endogenous adenosine, the concentration of adenosine in the media would have to be >1 μM in the cAMP assays and >0.5 μM in the [35S]GTP[S] assays in this study. Because these high concentrations of endogenous adenosine are unlikely, the results support the conclusion that the effects of PD 81,723 on forskolin-stimulated cAMP accumulation and [35S]GTP[S] binding are not mediated through the enhancement of the action of endogenous adenosine.
The results of the experiments provide substantial evidence that the agonist-independent effects of the allosteric enhancer observed in this study are neither mediated by the binding of PD 81,723 to the agonist binding site of the A1AdoR nor due to the potentiation by PD 81,723 of the actions of endogenous adenosine. The explanation for this unique effect of the allosteric enhancer may involve the overexpression of A1AdoR in this CHO cell line compared with the normal physiological level of receptor expression. For example, the results of saturation binding experiments using the A1AdoR antagonist [3H]CPX show that guinea pig forebrain tissue and these CHO cell membranes express ∼3000 (Kollias-Baker et al., 1994a) and 10,000 fmol/mg of protein of A1AdoR binding, respectively. Thus, the level of A1AdoR expression in the CHO cells is >3-fold higher than in guinea pig forebrain tissue. In other receptor systems (e.g., beta-2 adrenergic system), it has been hypothesized that a higher level of receptor expression increases the number of receptors coupled to G proteins, thereby permitting the detection of constitutive receptor activity (Bond et al., 1995; Lefkowitz et al., 1993; Milligan et al., 1995).
There is evidence in the results of our studies that A1AdoR expressed in these CHO cells exhibits constitutive receptor activity. The cellular cAMP accumulation in nontransfected cells was >2-fold higher than that in cells expressing the recombinant human A1AdoR. In addition, the A1AdoR antagonist CPX significantly increased cellular cAMP in CHO cells expressing recombinant A1AdoR but had no effect on cAMP accumulation in nontransfected cells (fig. 6). Consistent with these findings, baseline [35S]GTP[S] binding to CHO cell membranes expressing the recombinant human A1AdoR was significantly decreased by the A1AdoR antagonist CPX (fig. 7). Together, the results imply that in these cells, in the absence of an agonist, a significant number of A1AdoRs are coupled to Giproteins. CPX, acting as an inverse agonist, inhibits this constitutive receptor activity.
We hypothesize that the overexpression of A1AdoR in the CHO cells studied has enabled us to detect constitutive A1AdoR activity. PD 81,723 may potentiate the constitutive receptor activity by promoting isomerization of the A1AdoR to an active state of conformation. This explanation is in keeping with the idea that the fraction of receptors capable of interacting with G proteins in the absence of an agonist is dependent on a process of isomerization of receptors from an inactive to an active state that is governed by an equilibrium constant (Lefkowitz et al., 1993). Consistent with this theory, PD 81,723 has been shown to increase the fraction of A1AdoRs coupled to Gi proteins (Bhattacharya and Linden, 1995; Kollias-Baker et al., 1994a) and the binding of [35S]GTP[S] CHO cell membranes (fig. 7).
In summary, the results of this study show that in CHO cells stably expressing the recombinant human A1AdoR, the allosteric enhancer PD 81,723 alone, in the absence of an agonist, can attenuate forskolin-stimulated cAMP accumulation and increase [35S]GTP[S] binding. More importantly, these findings indicate that the allosteric enhancer does not mediate these effects by either binding to the agonist binding site of the A1AdoR or enhancing the action of endogenous adenosine present in the incubation media. These agonist-independent effects of PD 81,723 observed in CHO cells stably expressing the recombinant human A1AdoR may be due to enhancement of constitutive receptor activity.
Footnotes
-
Send reprint requests to: L. Belardinelli, M.D., University of Florida, Department of Medicine, P.O. Box 100277, Gainesville, FL 32610.
-
↵1 This work was supported by National Institutes of Health Grant HL-50488 (L.B.) and a postdoctoral fellowship from the American Heart Association, Florida Affiliate (C.A.K.-B.).
-
↵2 Current address: School of Veterinary Medicine, University of California, CVDLS, P.O. Box 1770, Davis, CA 95617.
- Abbreviations:
- PD 81
- 723, (2-amino-4,5-dimethyl-3-thienyl)-[3-(trifluromethyl)phenyl]methanone
- cAMP
- adenosine-3′,5′-cyclic monophosphate
- ADA
- adenosine deaminase
- GTPγS
- guanosine-5′-O-(3-thiotriphosphate)
- GDP
- guanosine-5′-diphosphate
- CPX
- 8-cyclopentyl-1,3-dipropylxanthine, CPA, N6-cyclopentyl adenosine
- (R)-PIA
- (R)-N6-(2-phenylisopropyl)adenosine
- CHA
- N6-cyclohexyladenosine
- HBSS
- Hanks’ balanced salt solution
- [3H]CPX
- 8-[dipropyl-2,3-3H(N)]cyclopentyl-1,3-dipropylxanthine
- [3H]CHA
- N6-[adenine-2,8-3H]cyclohexyladenosine
- [35S]GTP[S]
- [35S]-5′-(γ-thio)triphosphate
- [3H]cAMP
- [2,8-3H]adenosine-3′,5′-cyclic phosphate
- AdoR
- adenosine receptor
- G protein
- guanine nucleotide-binding protein
- CHO
- Chinese hamster ovary
- Received December 12, 1996.
- Accepted January 27, 1997.
- The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}