


Abstract
Dual-specificity protein phosphatase 5 (DUSP5) is a member of the tyrosine-threonine phosphatase family with the ability to dephosphorylate and inactivate extracellular signal-related kinase (ERK). The present study investigates whether knockout (KO) of Dusp5 improves renal hemodynamics and protects against hypertension-induced renal injury. The renal expression of DUSP5 was reduced, and the levels of phosphorylated (p) ERK1/2 and p-protein kinase C (PKC) α were elevated in the KO rats. KO of Dusp5 enhanced the myogenic tone of the renal afferent arteriole and interlobular artery in vitro with or without induction of deoxycorticosterone acetate-salt hypertension. Inhibition of ERK1/2 and PKC diminished the myogenic response to a greater extent in Dusp5 KO rats. Autoregulation of renal blood flow was significantly impaired in hypertensive wild-type (WT) rats but remained intact in Dusp5 KO animals. Proteinuria was markedly decreased in hypertensive KO versus WT rats. The degree of glomerular injury was reduced, and the expression of nephrin in the glomerulus was higher in hypertensive Dusp5 KO rats. Renal fibrosis and medullary protein cast formation were attenuated in hypertensive Dusp5 KO rats in association with decreased expression of monocyte chemoattractant protein 1, transforming growth factor-β1, matrix metalloproteinase (MMP) 2, and MMP9. These results indicate that KO of Dusp5 protects against hypertension-induced renal injury, at least in part, by maintaining the myogenic tone of the renal vasculature and extending the range of renal blood flow autoregulation to higher pressures, which diminish glomerular injury, protein cast formation, macrophage infiltration, and epithelial-mesenchymal transformation in the kidney.
Significance Statement Dual-specificity protein phosphatase 5 (DUSP5) is a tyrosine-threonine phosphatase that inactivates extracellular signal-related kinase (ERK). We previously reported that knockout (KO) of Dusp5 enhanced the myogenic response and autoregulation in the cerebral circulation. The present study investigates whether KO of DUSP5 improves renal hemodynamics and protects against hypertension-induced renal injury. Downregulation of DUSP5 enhanced the myogenic tone of renal arteriole and artery and autoregulation of renal blood flow in association with reduced proteinuria, glomerular injury, and interstitial fibrosis after the induction of hypertension. Inhibition of ERK1/2 and protein kinase C diminished the myogenic response to a greater extent in Dusp5 KO rats. These results suggest that DUSP5 might be a viable drug target for the treatment of hypertension nephropathy.
Introduction
Hypertension is the second leading cause of the end-stage renal disease (ESRD) after diabetes mellitus and accounts for about 45% of the ESRD cases worldwide (Bello et al., 2005). The prevalence of hypertension-induced chronic kidney disease is increasing (Bello et al., 2005; Webster et al., 2017), and more than 20% of hypertensive patients develop renal dysfunction in the United States (Collins et al., 2012). However, the genes and pathways that contribute to susceptibility to hypertensive nephropathy remain poorly understood. The Fawn-Hooded hypertensive (FHH) rat is a genetic model of hypertension-induced renal disease in which the myogenic reactivity and autoregulation of renal and cerebral blood flow (CBF) are impaired (van Dokkum et al., 1999; Mattson et al., 2007; Burke et al., 2013; Pabbidi et al., 2013). Transfer of a small region of chromosome 1 from Brown Norway (BN) rat onto an FHH.1BN congenic strain restores these responses and attenuates the development of renal disease (Mattson et al., 2005; Williams et al., 2011). Subsequent sequencing studies identified potential candidate genes in this region including Dual-specificity protein phosphatase 5 (Dusp5), a member of the tyrosine-threonine phosphatase family that dephosphorylates and inactivates extracellular signal-related kinase (ERK) (Owens and Keyse, 2007; Kucharska et al., 2009). Our previous study revealed that knockout (KO) of Dusp5 in an FHH.1BN congenic strain enhanced rather than inhibited the myogenic response of middle cerebral artery and autoregulation of CBF (Fan et al., 2014). However, the role of Dusp5 in the regulation of renal hemodynamics and its contribution to the development of hypertension-induced renal injury has not been studied. The present study examined the effects of the downregulation of DUSP5 on the myogenic reactivity of the afferent arteriole (Af-art) and interlobular artery (IA), autoregulation of renal blood flow (RBF), and their downstream renal pathologic consequences after the induction of deoxycorticosterone acetate (DOCA)-salt hypertension.
Materials and Methods
General.
Experiments were conducted in 10-week-old (n = 17) or 24-week-old male Dusp5 KO rats on the FHH.1BN congenic background (n = 106) that we previously generated (Fan et al., 2014). Heterozygous pairs were bred at the University of Mississippi Medical Center and the littermates of homozygous wild-type (WT) and Dusp5 KO rats were used to conduct all experiments. Ten-week-old rats were used to measure the myogenic response of Af-art to determine whether KO of Dusp5 in the absence of hypertension or renal injury altered renal vascular function. Mean arterial pressure (MAP) and protein excretion were examined from 21- to 24-week old animals. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
Changes of Arterial Pressure and Proteinuria in Response to DOCA-Salt Implantation.
Rats were maintained on the standard diet from birth to 21 weeks of age. The animals were treated with Baytril (10 mg/kg, s.c.) to prevent infection, the long-acting analgesic Rimadyl (5 mg/kg, s.c.) for pain relief prior to and the next morning after surgery, and anesthetized with isoflurane. A telemetry probe (HD-S10; Data Sciences International, St. Paul, MN) was implanted into the femoral artery, and the right kidney was removed. At the same time, a 21-day release DOCA (200 mg; Innovative Research of America, Sarasota, FL) or placebo pellet was implanted. After a 3-day recovery period, the baseline MAP was recorded between 10:00 AM and 2:00 PM for 3 days, and data were collected and analyzed using Ponemah software version 5.20 (Data Sciences International). Urine was collected overnight in metabolic cages. Total urinary protein was measured using the Bradford method with bovine serum albumin (BSA) as the standard (Bio-Rad, Hercules, CA). The rats were then given 1% sodium chloride (NaCl) drinking water to induce hypertension. MAP and proteinuria were measured on days 7, 14, and 21.
Autoregulation of RBF.
RBF autoregulation was determined as previously described (Roman and Cowley, 1985). Briefly, rats were anesthetized with ketamine (30 mg/kg, i.m.) and Inactin (50 mg/kg, i.p.) and were placed on a temperature-controlled surgical platform. Catheters were implanted in the carotid artery, femoral artery, and femoral vein for measurement of arterial pressure and intravenous infusions. Blood was collected at this time for measurement of plasma creatinine concentration. Two adjustable clamps were placed on the abdominal aorta above and below the left renal artery for altering renal perfusion pressure (RPP). A flow probe (Transonic Systems Inc., Ithaca, NY) was placed around the left renal artery for measuring RBF. The celiac and superior mesenteric arteries were ligated to adjust RPP to 150 mm Hg before running the autoregulatory curve. RBF was measured over a range of RPP from 150 to 60 mm Hg in steps of 10 mm Hg with a 3-minute equilibration period between each point by tightening the clamp above the renal artery. Then, RPP was adjusted back to 150 mm Hg by releasing this clamp, and RBF was measured over a range of RPP from 150 to 180 mm Hg by tightening the clamp below the renal artery while pressure was measured from the carotid artery. At the end of the experiment, all clamps were released and blood pressure returned to approximately 150 mm Hg. The kidney was harvested and snap frozen in liquid nitrogen or was rapidly immersed in 10% neutral buffered formalin for Western blot or immunohistochemistry.
Myogenic Reactivity of Renal Af-art and IA.
Af-art with an attached glomerulus was microdissected from 10-week-old WT (n = 11) and Dusp5 KO (n = 6) rats, and was cannulated with concentric glass pipettes as previously described (Ge et al., 2014; Fan et al., 2017). The diameter of the Af-art was measured at perfusion pressures of 60 and 120 mm Hg. Then, the solution in the bath was replaced with calcium-free physiologic salt solution and the response was redetermined.
Renal IA was dissected from 24-week WT and Dusp5 KO rats (total, 29 rats) (for details, see Figs. 2B and 3) with or without DOCA-salt treatments. The vessels were mounted on glass micropipettes, and the diameters were measured at perfusion pressures of 60, 120, and 180 mm Hg in the presence or absence of calcium (Fan et al., 2015b) before and after the addition of a protein kinase C (PKC) inhibitor Bisindolylmaleimide III (BIM III; 300 nM; Santa Cruz Biotechnology, Dallas, TX) or an ERK inhibitor, FR180204 (1 μM; Santa Cruz Biotechnology), to the bath. The same volume of vehicle (DMSO, 0.1%) was applied in the bath as a control for BIM III or FR180204.
Glomerular Permeability to Albumin.
A separate group of rats (n = 16) were anesthetized and given intravenous injection of high-molecular-weight (500 kDa) fluorescein isothiocyanate-dextran in 0.9% NaCl solution. Glomeruli were isolated by passing minced renal cortex through filters (U.S.A. standard test sieves No. 100 and No. 140; Thermo Fisher Scientific, Waltham, MA) and were captured on a 70-μm cell strainer (BD Biosciences, San Jose, CA). The isolated glomeruli were placed on a coverslip coated with poly-l-lysine (10 mg/ml; Sigma-Aldrich, St. Louis, MO), superfused with ice-cold Hanks’ balanced salt solution containing 6% BSA, and imaged as we previously reported (Fan et al., 2015a). Baseline fluorescence intensity was recorded, then the bath was switched to 4% BSA and the changes in fluorescence intensity were measured. The reflection coefficient (σ) was calculated as the ratio of the change in fluorescence intensity to the expected change (33%), and the convectional permeability to albumin calculated as 1 − σ, as previously described (Savin et al., 1992; Fan et al., 2015a) as an index of glomerular permeability to albumin (Palb) to compare the relative degree of glomerular injury between normotensive and hypertensive Dusp KO and WT rats.
Histology and Immunohistochemistry.
Paraffin sections (5 µm) were prepared and stained with Masson’s trichrome or H&E to evaluate the degree of glomerular injury and renal fibrosis as previously described (Muroya et al., 2015). The areas of protein casts stained red on H&E sections were analyzed. The sections were immunostained for nephrin (Fitzgerald, Washington, DC), TGF-β1 (Promega, Madison, WI), and cluster of differentiation 68 (ED-1) (CD68; Bio-Rad). Kidney tissue sections were deparaffinized with xylene for 5 minutes and rehydrated in 99.5%, 90%, 80%, and 70% ethanol for 5 minutes, respectively. The sections were permeabilized in 0.1% trypsin buffer for 30 minutes at 37°C followed by antigen retrieval in citrate buffer, heated to 98°C for 20 minutes, and rinsed with cold methanol. Sections were incubated at 4°C with antibodies to TGF-β1 (1:200), nephrin (1: 50), or ED-1 (1:100) overnight, then were incubated with Alexa Fluor–labeled secondary antibodies for 1 hour at room temperature. The sections were counterstained with 0.005% Evans blue to quench endogenous fluorescence, washed, and coverslipped using an antifade mounting medium containing Hoechst stain. Nephrin expression was quantified as the percentage of fluorescence staining in each glomerulus. ED-1 expression was calculated as the numbers of infiltrating macrophages per glomerulus. Ten glomeruli in each section were measured, and six sections from 6 to 10 rats per group were counted.
Western Blotting.
Proteins extracted from renal cortex were separated on an SDS-polyacrylamide gel, transferred to a 0.2-μm nitrocellulose membrane, blocked with 5% milk, and incubated with antibodies to β-actin (Abcam, Cambridge, MA), monocyte chemoattractant protein 1 (MCP-1) (Thermo Fisher Scientific), TGF-β1 (Santa Cruz Biotechnology), matrix metalloproteinase (MMP) 2 (Novus, Littleton, CO), MMP9 (Abcam), ERK1/2 (Cell Signaling Technology, Danvers, MA), phosphorylated (p)-ERK1/2 (Cell Signaling Technology), PKCα (Santa Cruz Biotechnology), p-PKCα (Santa Cruz Biotechnology), DUSP5 (Abnova, Littleton, CO), α-smooth muscle actin (α-SMA; Sigma-Aldrich), E-cadherin (BD Biosciences), or vimentin (Cell Signaling Technology) followed by horseradish peroxidase–coupled secondary antibodies. The blots were developed using SuperSignalR ECL reagent (Thermo Fisher Scientific) and analyzed using a Bio-Rad Chemidoc XRS+ Imaging System (N = 4–6 rats per group). Experiments were repeated two to three times.
Statistical Analysis.
Mean values ± S.E.M. are presented. The significance of differences in mean values between groups was determined using a two-way ANOVA (strain, treatment) and a two-way ANOVA for repeated measures followed by a Holm-Sidak test. A value of P < 0.05 was considered to be significant.
Results
Validation of KO of Dusp5 in the Kidney.
We previously created a global Dusp5 KO rat strain with the congenic FHH.1BN genetic background and reported that KO of Dusp5 enhanced the myogenic response and autoregulation of CBF (Fan et al., 2014). The present study explored the role of the downregulation of Dusp5 on renal hemodynamics and its contribution to the development of hypertension-induced renal injury. The results shown in Fig. 1A confirmed that the expression of DUSP5 protein was markedly reduced in the renal cortex of normotensive or hypertensive Dusp5 KO rats relative to WT controls. This observation was consistent with what we found in the brain, liver, spleen, cerebral vessels, and white blood cells (Fan et al., 2014), and is associated with increased renal expression of p-ERK1/2, the primary substrate of DUSP5 (Fig. 1B), and p-PKCα (Fig. 1C). There was no difference in body weight between WT and Dusp5 KO rats prior to (299 ± 5 and 296 ± 7 g, respectively; n = 12 of each strain) and after (305 ± 6 and 302 ± 12 g, respectively; n = 9 of each strain) DOCA/salt treatment. Similarly, baseline kidney weight was not different between WT and Dusp5 KO rats (1.21 ± 0.05 and 1.18 ± 0.06 g, respectively; n = 12 of each strain); however, it was greater in WT rats compared with Dusp5 KO rats after the induction of hypertension (3.4 ± 0.1 and 2.9 ± 0.1 g, respectively; n = 9 of each strain).

Validation of KO of Dusp5 in the kidney. (A) Representative images of Western blots and quantification of DUSP5 expression in renal cortical homogenates from normotensive and DOCA-salt hypertensive Dusp5 KO and WT rats. (B) Representative images of Western blots and quantification of the ratio of p-ERK1/2 to ERK1/2 in the renal cortex. (C) Representative images of Western blots and quantification of the ratio of phosphorylated to total PKCα in renal cortical homogenates. Mean intensities relative to the values obtained in normotensive WT rats ± S.E.M. are presented. N = 4–6 rats per group. Experiments were repeated two to three times. *P < 0.05 from the corresponding value in Dusp5 KO rats vs. WT.
Effects of KO of Dusp5 on Renal Hemodynamics.
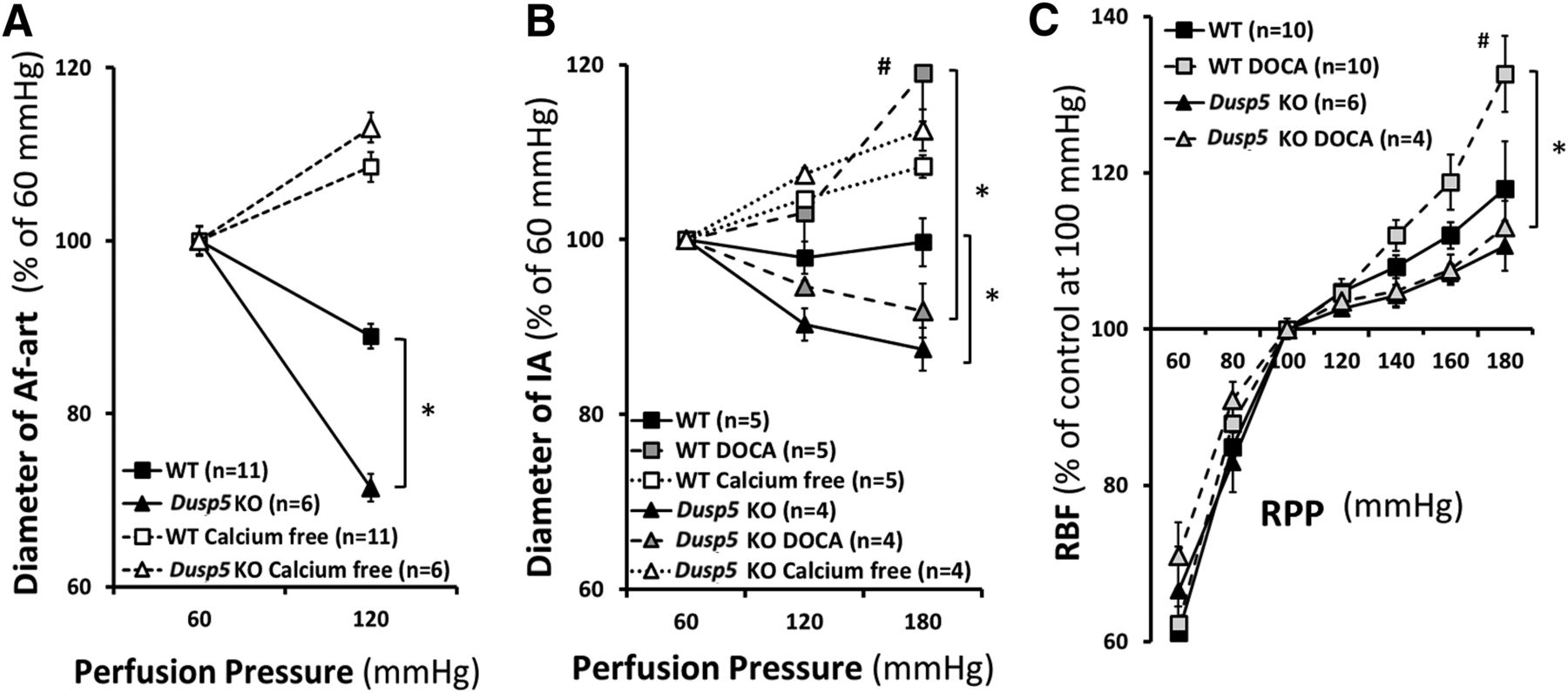
Baseline diameters of the Af-art and IA were similar in 10- or 24-week-old Dusp5 KO and WT rats at a perfusion pressure of 60 mm Hg. As shown in Fig. 2 and Table 1, the diameter of the Af-art decreased by 29% ± 2% in Dusp5 KO rats when the perfusion pressure increased from 60 to 120 mm Hg, whereas it only fell by 11% ± 1% in WT rats. The luminal diameter of the IA also decreased to a greater extent in 24-week-old placebo-treated Dusp5 KO rats compared with WT rats when the perfusion pressure increased from 60 to 180 mm Hg. These vessels still constricted in response to elevations in pressure in hypertensive Dusp5 KO rats, whereas they lost the ability to constrict and dilated in DOCA-salt–treated WT animals. There was no significant difference in passive diameters of Af-art and IA measured in the calcium-free medium in normotensive or hypertensive Dusp5 KO and WT rats.
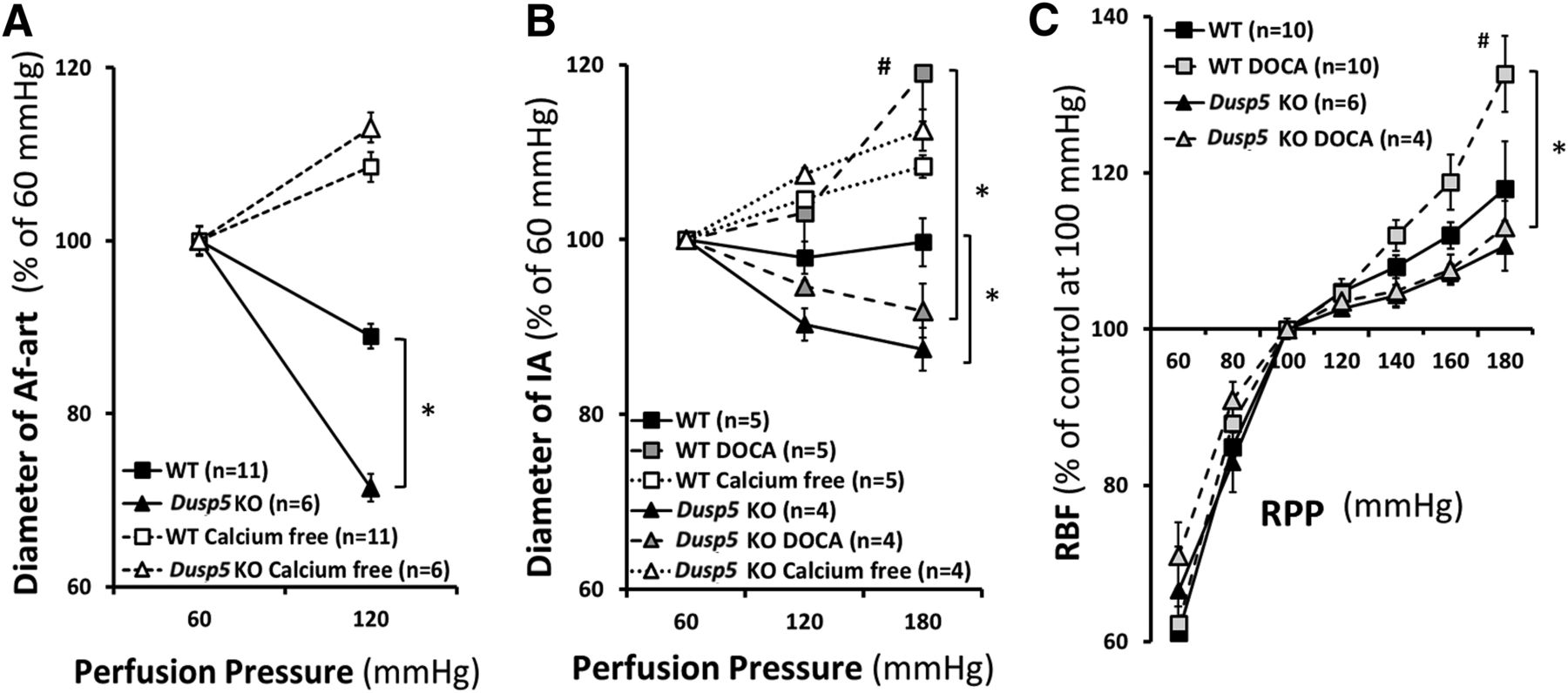
Effects of Dusp5 KO on renal hemodynamics. (A) Comparison of the changes in the diameter of isolated perfused Af-arts of 10-week-old normotensive rats in response to an increase in perfusion pressure from 60 to 120 mm Hg before and after removal of calcium from the bath. (B) Comparison of the myogenic response of isolated renal IAs of 24-week-old normotensive and DOCA-salt hypertensive rats in response to an increase in perfusion pressure from 60 to 180 mm Hg. (C) Comparison of autoregulation of RBF in 24-week-old normotensive and hypertensive rats. Mean values ± S.E.M. are presented. Numbers in parentheses indicate the number of animals studied per group. *P < 0.05 from the corresponding value in Dusp5 KO vs. WT rats. #P < 0.05 from DOCA-salt treated vs. baseline value within strain.
Inner diameters of isolated perfused afferent arteriole and interlobular artery in response to elevations in perfusion pressure
Comparison of absolute values of inner diameter of isolated perfused Af-art and IA in response to elevations in perfusion pressure. Mean values ± S.E.M. are presented.
A comparison of autoregulation of RBF in Dusp5 KO and WT rats is presented in Fig. 2C and Table 2. Baseline RBF was similar in Dusp5 KO rats (7.5 ml/min) and WT rats (7.2 ml/min) measured at an RPP of 100 mm Hg. RBF was well autoregulated (<10% change) in both normotensive Dusp5 KO and WT rats over the normal autoregulated range of pressures from 80 to 140 mm Hg. However, autoregulation was still well maintained in Dusp5 KO rats when challenging with acute or chronic hypertension, and RBF only increased by 11% ± 3% and 13% ± 1%, respectively, when MAP was increased from 100 to 180 mm Hg. In contrast, autoregulation was impaired in hypertensive WT rats, and RBF rose by 33% ± 5% over the same range of pressure. The autoregulatory breakthrough point was also shifted to lower pressures in WT rats, especially after the induction of hypertension.
Absolute values of renal blood flow in response to changes in renal perfusion pressure
Comparison of absolute values of RBF in response to changes in RPP. Mean values ± S.E.M. are presented.
Effects of Inhibition of ERK1/2 and PKC on Renal Myogenic Reactivity.
We compared the myogenic reactivity of renal IA in response to a PKC inhibitor BIM III (IC50 = 26 nM), and FR180204 that selectively inhibits ERK1 (IC50 = 510 nM) and ERK2 (IC50 = 300 nM). We first obtained time and dose curves of these inhibitors (data not shown) and determined treatment of the vessels with BIM III (300 nM) and FR180204 (1 μM) for 30 minutes compared with vehicle (DMSO)-treated control vessels. We found that the inhibition of ERK1/2 and PKC diminished myogenic reactivity and increased the diameter of IA to a significantly greater extent in Dusp5 KO rats than WT rats at all pressures studied (Fig. 3; Table 3). These results indicate that the activation of ERK and PKC signaling pathways contribute to the enhanced myogenic reactivity in the renal vasculature in Dusp5 KO rats.

Effects of inhibition of ERK1/2 and PKC on the myogenic reactivity of interlobular artery. The percentage of changes of inner diameters were compared in response to an increase in perfusion pressure from 60 to 180 mm Hg in the presence or absence of a selective ERK1/2 inhibitor, FR180204, and a selective PKC inhibitor, BIM III, in isolated renal IAs of 24-week-old normotensive rats. Mean values ± S.E.M. are presented. Numbers in parentheses indicate the number of animals studied per group. *P < 0.05 from the corresponding value in Dusp5 KO vs. WT rats. #P < 0.05 from the changes in diameter in response to drug treatments within strain.
Inner diameters of isolated perfused interlobular artery in response to selective ERK1/2 inhibitor FR180204 and PKC inhibitor BIM III
Absolute values of inner diameter of isolated perfused IA and AA in the presence of a selective ERK1/2 inhibitor FR180204 (1 μM) and PKC inhibitor BIM III. Mean values ± S.E.M. are presented.
Time Courses of the Development of Hypertension and Proteinuria.
As presented in Fig. 4, baseline MAP was similar (115 mm Hg) in normotensive Dusp5 KO and WT rats. MAP was significantly increased after 3 weeks of DOCA-salt treatment, which was indistinguishable between hypertensive Dusp5 KO (169 ± 5 mm Hg) and WT (177 ± 5 mm Hg) rats. In contrast, the rise in urinary protein excretion was significantly attenuated in hypertensive Dusp5 KO rats (226 ± 24 mg/day) compared with WT rats (512 ± 23 mg/day). This was associated with reduced serum creatinine levels in hypertensive Dusp5 KO versus WT rats [0.56 ± 0.04 vs. 0.82 ± 0.10 mg/dl (n = 8), respectively], whereas there was no difference between these two strains implanted with placebo pellets.
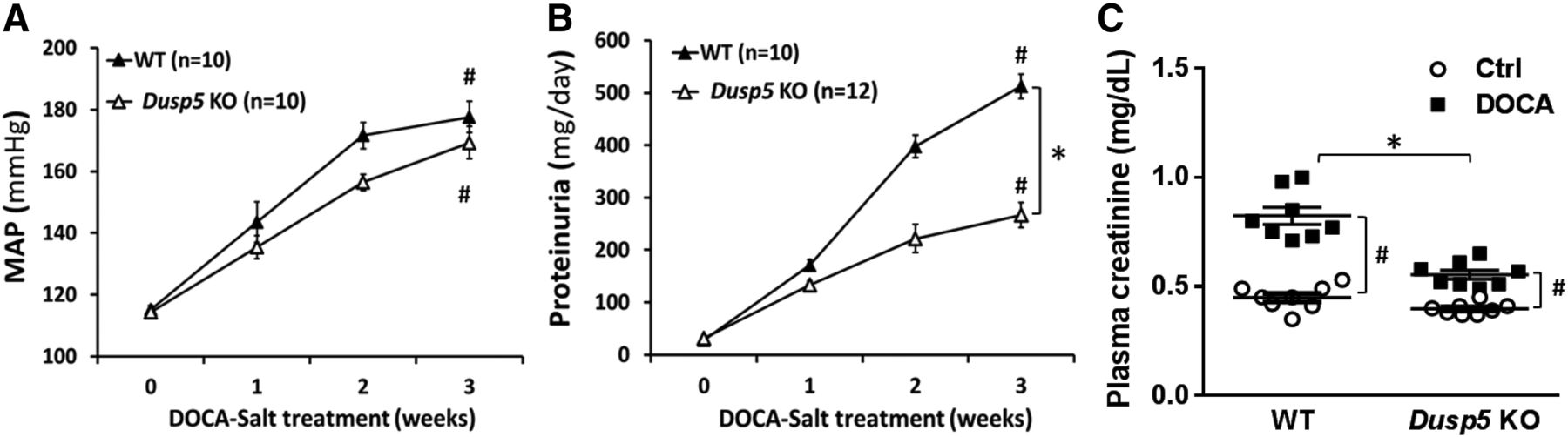
Effect of Dusp5 KO on the development of hypertension and proteinuria. The time courses of changes in MAP (A) and proteinuria (B) after the induction of DOCA-salt hypertension in WT and Dusp5 KO rats. (C) Comparison of plasma creatinine concentration after the induction of DOCA-salt hypertension in WT and Dusp5 KO rats (n = 8 rats in each group). Mean values ± S.E.M. are presented. Numbers in parentheses indicate the number of animals studied per group. *P < 0.05 from the corresponding value in Dusp5 KO rats vs. WT rats. #P < 0.05 from DOCA-salt treated value vs. control within strain.
Glomerular Injury, Renal Interstitial Fibrosis, Tubular Injury, and TGF-β/MMPs Expression.
The effects of KO of Dusp5 on renal injury were explored by comparing glomerular injury score, nephrin expression, Palb, renal interstitial fibrosis, protein cast formation, and TGF-β1/MMPs expression in normotensive and hypertensive Dusp5 KO and WT rats. Baseline glomerular injury scores were similar in Dusp5 KO and WT rats. However, the glomerular injury score was significantly reduced in Dusp5 KO rats compared with WT rats after 3 weeks of hypertension (2.2 ± 0.1 and 3.1 ± 0.1, respectively) (Fig. 5A). Nephrin was uniformly expressed in the glomeruli of Dusp5 KO and WT rats prior to the development of hypertension. After 3 weeks of DOCA-salt treatment, the glomerular nephrin expression fell to a greater extent in the glomeruli of hypertensive WT than Dusp5 KO rats, which is consistent with the loss of podocytes in the KO strain (Fig. 5B). The convectional permeability to albumin, an index of Palb, was similar in normotensive Dusp5 KO and WT rats. It remained relatively normal in Dusp5 KO rats, while it significantly increased in WT rats after the induction of hypertension (Fig. 5C).

Effects of Dusp5 KO on glomerular injury. (A) Comparison of glomerular injury in the renal cortex of normotensive and hypertensive WT and Dusp5 KO rats. (B) Representative pictures of nephrin immunostaining in glomeruli of normotensive and hypertensive Dusp5 KO and WT rats. (C) Comparison of Palb in isolated glomeruli. The decline in fluorescence intensity was greater in the glomeruli of DOCA-salt–treated Dusp5 KO and WT rats, indicating an increased reflection coefficient (σ). N = 4–6 rats per group. Numbers in the parentheses indicate the numbers of rat and glomeruli studied. Mean values ± S.E.M. are presented. *P < 0.05 from the corresponding value in Dusp5 KO vs. WT rats. #P < 0.05 from DOCA-salt treated vs. baseline value within strain.
The degree of renal interstitial fibrosis was very low in Dusp5 KO than WT rats prior to the induction of hypertension. Renal interstitial fibrosis increased in both strains, but it was significantly lower in Dusp5 KO than WT rats after the development of hypertension (Fig. 6A). Similarly, the formation of protein casts in the outer medulla was reduced in hypertensive Dusp5 KO (10% ± 0.1%) rats compared with WT (24% ± 0.5%) controls, though there was no difference before the induction of hypertension (Fig. 6B).

Effects of Dusp5 KO on renal interstitial fibrosis, tubular injury, and TGF-β/MMPs expression. (A) Comparison of renal interstitial fibrosis in normotensive and hypertensive Dusp5 KO and WT rats. The slides were stained with Masson’s trichrome, and the degree of fibrosis (blue color) was measured by thresholding the percentage area of blue color per field. (B) Comparison of protein cast formation in the outer medulla of normotensive and hypertensive WT and Dusp5 KO rats. The slides were stained with H&E, and the area of protein casts was measured by thresholding the percentage of area occupied by eosin red fluorescence per field. (C) Representative pictures of TGF-β1 immunofluorescent staining in the renal cortex in normotensive and hypertensive Dusp5 KO and WT rats. (D–F) Comparison of TGF-β1, MMP2, and MMP9 expression in the renal cortex of normotensive and hypertensive Dusp5 KO and WT rats by Western blot. N = 4–6 rats per group. Experiments were repeated two to three times. Mean values ± S.E.M. are presented. *P < 0.05 from the corresponding value in Dusp5 KO vs. WT rats. #P < 0.05 from DOCA-salt treated vs. baseline value within strain.
The expression of TGF-β1 was barely detectable in the kidneys of normotensive Dusp5 KO and WT rats by immunofluorescence staining and Western blotting. The expression of TGF-β1 increased in the proximal tubule of both Dusp5 KO and WT rats after the induction of hypertension, but the increase was less in Dusp5 KO rats than in WT rats (Fig. 6C). TGF-β1 expression was lower in Dusp5 KO than WT rats after the development of hypertension (Fig. 6D). The expression of MMP2 and MMP9 was similar in the kidneys of normotensive Dusp5 KO and WT rats. MMP2 and MMP9 expression increased in both strains after the development of hypertension. However, the expression of MMP2 and MMP9 remained lower in the kidneys of hypertensive Dusp5 KO rats than in those of the WT controls (Fig. 6, E and F).
Effects of Dusp5 KO on Expression of MCP-1, Cluster of Differentiation 68, and Biomarkers of Epithelial-Mesenchymal Transition.
The expression of MCP-1 was lower in hypertensive Dusp5 KO rats (Fig. 7A) in association with reduced infiltration of the ED-1-positive macrophages in the kidney (Fig. 7B). In addition, the expression of mesenchymal phenotype biomarkers vimentin and α-SMA were reduced and the expression of epithelial biomarker E-cadherin was increased in hypertensive Dusp5 KO as compared with hypertensive WT rats (Fig. 8).

Expression of MCP-1 and ED-1. (A) Comparison of MCP-1 expression in the renal cortex of WT and Dusp5 KO rats relative to the mean control value in normotensive WT rats. (B) Representative images of ED-1 immunofluorescent staining and quantification of ED-1–positive infiltrating macrophages in the renal cortex of WT and Dusp5 KO rats. Yellow arrows point to the ED-1–positive macrophages. Mean values ± S.E.M. are presented. *P < 0.05 from the corresponding value in Dusp5 KO vs. WT rats. #P < 0.05 from DOCA-salt treated vs. baseline value within strain.

Expression of biomarkers of epithelial-mesenchymal transition. Comparison of the expression of EMT biomarkers: E-cadherin, α-SMA, and vimentin in the renal cortex of WT and Dusp5 KO rats. Mean values ± S.E.M. are presented. *P < 0.05 from the corresponding value in Dusp5 KO vs. WT rats. #P < 0.05 from DOCA-salt treated vs. baseline value within strain.
Discussion
DUSP5 is a member of the tyrosine-threonine phosphatase family that dephosphorylates and inactivates p-ERK1/2 and p-PKC (Owens and Keyse, 2007; Fan et al., 2014). We have reported that KO of Dusp5 enhances the myogenic response of cerebral arteries and extends the range of CBF autoregulation to higher pressures (Fan et al., 2014). The present study investigated whether KO of Dusp5 also improves renal hemodynamics and protects against hypertension-induced renal injury.
We first confirmed that the expression of DUSP5 was diminished in the kidney in this global Dusp5 KO strain, similar to what we reported in other tissues (Fan et al., 2014), in association with increased expression of p-ERK1/2 and p-PKCα. ERK1 and ERK2 are the primary substrates of DUSP5 and can be dephosphorylated by this phosphatase. Increases in phosphorylation of ERK1/2 and PKC in vascular smooth muscle cells (VSMCs) contribute to enhanced vascular reactivity by activation of transient receptor potential and to blockade of BK channels to promote calcium influx in response to elevations in transmural pressure, and the administration of angiotensin II (AngII) and other vasoactive agonists (Sun et al., 1999; Murphy et al., 2002; Zhao et al., 2005; Fan et al., 2013; Earley and Brayden, 2015). Increased intracellular calcium levels lead to activation of myosin light chain (MLC) kinase, which enhances phosphorylation of MLCs causing vasoconstriction (Roberts, 2012). Activated ERK also phosphorylates caldesmon, an actin-binding protein, to diminish its inhibitory effect on myosin-ATPase to VSMC constriction (Ngai and Walsh, 1984). PKC phosphorylates the inhibitory protein of MLC phosphatase CPI-17 to increase the MLC phosphorylation contributing to VSMC constriction (Woodsome et al., 2001). The enhanced constrictive capability of VSMCs also could be due to that PKCα phosphorylates actin-associated regulatory protein calponin (Kanashiro and Khalil, 1998). We found that inhibition of ERK1/2 and PKC diminished the vascular constriction of renal IA to a significantly greater extent in Dusp5 KO rats than WT rats in response to pressure, suggesting that the activation of ERK and PKC signaling pathways contributes to the enhanced myogenic reactivity in Dusp5 KO rats. Consistent with this view, we found that KO of Dusp5 increased the myogenic response of isolated perfused Af-art similar to what we reported in middle cerebral arteries (Fan et al., 2014). In addition, the luminal diameter of the IA reduced to a greater extent in 24-week normotensive Dusp5 KO compared with WT rats when the perfusion pressure increased from 60 to 180 mm Hg. These vessels retained responsiveness to pressure in hypertensive Dusp5 KO rats, but the myogenic tone was diminished in hypertensive WT animals. These results demonstrate that downregulation of DUSP5 expression increases vascular reactivity in response to pressure in both of the Af-art (∼20 μm) and IA (∼50 μm).
After challenged with DOCA-salt hypertension, IA of Dusp5 KO rats maintained intact myogenic tone, and RBF was well autoregulated up to 180 mm Hg. In contrast, RBF autoregulation was intact in normotensive WT rats but was impaired at high pressures after DOCA-salt hypertension. This could be, in part, due to the resetting/suppression of tubuloglomerular feedback, diminishing the myogenic response of renal arterioles/arteries secondary to volume expansion seen in the DOCA-salt hypertensive model (Dworkin et al., 1984; Holstein-Rathlou et al., 1991; Moreno et al., 2001; Bae et al., 2009). The enhanced myogenic tone in the Dusp5 KO model preserved the impaired RBF autoregulation and protected against hypertension-induced renal injury.
Autoregulation of RBF is a critical homeostatic mechanism that normally protects the glomerulus from damage after elevations in pressure (Navar, 1978; Loutzenhiser et al., 2006; Burke et al., 2014; Carlstrom et al., 2015; Just, 2007). It is able to maintain blood flow to the kidney or brain to a pressure about 30%–40% above resting pressures (Cipolla, 2010). An autoregulation breakthrough point occurs at perfusion pressure above the upper limit of the myogenic response, at which point vascular resistance is reduced by forced dilation of the vessels. We found that the autoregulatory breakthrough point was shifted to lower pressures in hypertensive WT rats, and the myogenic tone and RBF autoregulation remained intact in Dusp5 KO DOCA rats, indicating that the downregulation of DUSP5 plays a renal protective role. Impaired RBF autoregulation increases the transmission of pressure to the glomerulus in volume-expanded models of hypertension, such as Dahl salt-sensitive (Roman, 1986), DOCA-salt (Moore et al., 1979), and reduced renal mass hypertensive models (Bidani et al., 1987; Brown et al., 1995; Carlstrom et al., 2015). These models exhibit marked hypertrophy of glomeruli and rapidly develop proteinuria, progressive focal glomerulosclerosis, and severe renal interstitial fibrosis (Moore et al., 1979; Roman, 1986; Bidani et al., 1987; Brown et al., 1995). Autoregulation of RBF is intact on enhanced in spontaneously hypertensive rat and Ang II–infused models of hypertension. Elevated preglomerular vascular resistance that protects the glomerulus from increases in systemic pressure is thought to underlie the resistance to proteinuria and renal injury (Iversen et al., 1987; Arendshorst et al., 1999; Carlstrom et al., 2015). These models typically exhibit less glomerular hypertrophy, glomerular sclerosis, and renal interstitial fibrosis than those hypertensive models with impaired autoregulation. Moreover, the degree of renal injury in spontaneously hypertensive rat and AngII models is less severe and is associated with renal ischemia and inflammation (Navar, 2014).
Renal hypertrophy is an adaptive mechanism that accelerates hypertension-induced renal injury. In this study, we found that there was greater kidney hypertrophy in WT DOCA rats, and they developed more severe proteinuria and renal injury than Dusp5 KO animals. Although we do not have direct evidence, it is unlikely that DUSP5 promoted renal growth in hypertensive WT rats since it reduced phosphorylation of ERK1/2 and PKCα, which enhanced cell proliferation (Chambard et al., 2007; Gao et al., 2009).
The mechanisms by which hypertension promotes the development of renal injury remain to be fully elucidated. Indeed, increased transmission of pressure to the glomerulus in association with podocyte loss and foot process effacement, and increased glomerular permeability to protein play an important role in hypertension-induced ESRD (Neuringer and Brenner, 1993; Ritz et al., 1993; Karlsen et al., 1997; Bidani et al., 2003; Bidani and Griffin, 2004). The present finding that the expression of nephrin in the kidney and glomerulus was markedly decreased in hypertensive WT rats was consistent with the loss of podocytes. In contrast, nephrin expression was better maintained in the Dusp5 KO animals in which RBF was autoregulated to higher pressures. Palb also increased in glomeruli isolated from hypertensive WT rats, but remained relatively unchanged in Dusp5 KO animals. Moreover, the degree of proteinuria was twice as high in hypertensive WT animals than in Dusp5 KO animals, even though blood pressures were not significantly different in the two groups.
Convectional permeability (1 − σ) was used as an index of Palb since it reflects the water movement directly proportional to the applied oncotic gradients (albumin concentration gradients), which is distinct from diffusional permeability that is independent of solvent movement (Savin et al., 1992). We recognize that 1 − σ determined by our fluorescence dilution technique was higher than the diffusive permeability to albumin across individual capillary estimated by other techniques. However, this method could provide essential information to compare the relative degree of injury in a large population of glomeruli.
The loss of podocytes and glomerular hypertrophy after uninephrectomy and induction of DOCA-salt hypertension increased the single-nephron glomerular filtration rate and elevated the filtration of protein through the glomerular filtration barrier triggers the development of proteinuria, as is often seen in diabetic nephropathy (Dworkin et al., 1984; Kretzler et al., 1994; Langham et al., 2002). Accelerated delivery of protein to the proximal tubule initially produces a compensatory increase in protein reuptake (Abbate et al., 2006). Glomerular mesangial cells and tubular epithelial cells challenged with protein overload are damaged and produce inflammatory cytokines and profibrotic mediators, such as MCP-1, leading to recruitment and accumulation of monocytes/macrophages in glomeruli and the interstitium (Wang et al., 1997, 1999; Donadelli et al., 2000; Eardley et al., 2006). In the present study, we found that KO of Dusp5 was associated with downregulated expression of MCP-1 and reduced infiltration of macrophages (seen as ED-1 positive) in the kidney, and decreased glomerulosclerosis and renal interstitial fibrosis. We also found that the renal expression of MMP2 and MMP9 increased after the development of hypertension, but the levels were much higher in the WT than in Dusp5 KO rats. MMP2 and MMP9 play an antifibrotic role in renal fibrosis due to proteolytic degradation of extracellular matrix (Hijova, 2005). They are also implicated in the initiation and progression of kidney fibrosis by releasing active TGF-β1 from its latent complex as well as activating TGF-β1 type II receptor (Yu and Stamenkovic, 2000; Wang et al., 2006; Zhao et al., 2013). They are also necessary and sufficient to promote epithelial-mesenchymal transition (EMT) in proximal tubule cells and to activate resident myofibroblasts in the kidney (Lenz et al., 2000; Cheng and Lovett, 2003; Liu, 2004; Cheng et al., 2006; Catania et al., 2007; Zhao et al., 2013). KO of Dusp5 was associated with reducing the EMT process, as indicated by decreased expression of mesenchymal phenotype biomarkers (e.g., vimentin and α-SMA) and increased expression of epithelial biomarker (e.g., E-cadherin) compared with hypertensive WT rats. Thus, our results indicate that KO of Dusp5 protects against renal fibrosis through the inhibition of fibrogenesis via an MCP-1/macrophage/TGF-β1/MMPs/EMT–involved mechanism.
As an essential modulator of mitogen-activated protein kinase, DUSP5 plays important roles in vascular development and function (Pramanik et al., 2009; Nayak et al., 2014), ischemic stroke (Mengozzi et al., 2012), cancer (Ueda et al., 2003; Pratilas et al., 2009; Yun et al., 2009; Montero-Conde et al., 2013; Shin et al., 2013; Rushworth et al., 2014; Wang et al., 2019), and the immune system (Holmes et al., 2015; Kutty et al., 2016; Manley et al., 2019), suggesting that it could be an attractive drug target. However, based on the nuclear localization, the similarity of the structure and active sites of DUSP isoforms, as well as the interaction with ERK, it has been difficult to develop specific inhibitors of DUSP5. Two classes of DUSP5 inhibitors have been described: compounds contain or are similar to naphthalene trisulfonate core, such as suramin, a Food and Drug Administration–approved drug for the treatment of African sleeping sickness, and a small molecule, CSD3-2320, that binds to the C-terminal phosphatase domain of DUSP5 to block its phosphatase effect (Neumann et al., 2015). More recently, halogenated xanthene inhibitors of DUSP5 were discovered (Bongard et al., 2019). These xanthene-ring analogs inhibit DUSP5 phosphatase domain activity in vitro with IC50 values in the micromolar range and relative potency of rose bengal > merbromin > erythrosin B > eosin Y. To date, none of the DUSP5 inhibitors have completed preclinical trials for cancer, immune disorders, or vascular dysfunction and entered clinical trials. Although the present study suggests that increases in ERK and PKC activity in Dusp5 KO rats enhanced the myogenic reactivity in the renal circulation and afforded renoprotection in DOCA-salt hypertension, one argument of the use of DUSP5 inhibitors to protect the kidney from hypertension is that they might increase the severity of hypertension by also increasing resistance in other vascular beds. This does not seem to be the case in the present study since blood pressure was similar in WT and Dusp5 KO rats under control conditions and after DOCA/salt treatment. However, additional preclinical work will be needed to access the potential renoprotective actions of DUSP5 inhibitors in other models.
In conclusion, the present study demonstrates that KO of Dusp5 is associated with activation of PKCα and ERK1/2, enhanced the myogenic reactivity of Af-art and preglomerular arteries, and improved RBF autoregulation to higher pressures after the induction of DOCA-salt hypertension. This is associated with decreased glomerular injury and loss of podocytes, reduced glomerular permeability, and proteinuria. KO of Dusp5 also reduced the expression of MCP-1, decreased the infiltration of macrophages and downregulation of the expression of TGF-β1, MMP2, and MMP9 in the kidney, which reduced EMT and renal fibrosis. Taken together, the inhibition of Dusp5 could protect against hypertension induced renal injury, at least in part, by enhancing renal vascular reactivity and by attenuating the transmission of elevated pressure to the glomerulus.
Authorship Contributions
Participated in research design: Roman and Fan.
Conducted experiments: C. Zhang, He, Murphy, H. Zhang, Wang, Ge, Gao, Geurts, and Fan.
Performed data analysis: C. Zhang, He, Geurts, Roman, and Fan.
Wrote or contributed to the writing of the manuscript: C. Zhang, Murphy, Williams, Geurts, Roman, and Fan.
Footnotes
- Received April 15, 2019.
- Accepted May 16, 2019.
This study was supported by the National Institutes of Health (Grants AG-050049 and AG-057842 to F.F.; P20-GM-104357 to J.M.W., R.J.R., and F.F.; DK-109133 to J.M.W.; and HL-36279, DK-104184 and HL-138685 to R.J.R.), the American Heart Association (Grant 16GRNT31200036 to F.F.), and the China Scholarship Council (Fellowship 201606100208 to C.Z.).
Abbreviations
- Af-art
- afferent arteriole
- α-SMA
- α-smooth muscle actin
- AngII
- angiotensin II
- BIM III
- bisindolylmaleimide III
- BN
- Brown Norway
- BSA
- bovine serum albumin
- CBF
- cerebral blood flow
- DOCA
- deoxycorticosterone acetate
- DUSP5
- dual-specificity protein phosphatase 5
- ED-1
- cluster of differentiation 68
- EMT
- epithelial-mesenchymal transition
- ERK
- extracellular signal-related kinase
- ESRD
- end-stage renal disease
- FHH
- Fawn-Hooded hypertensive
- IA
- interlobular artery
- KO
- knockout
- MAP
- mean arterial pressure
- MCP-1
- monocyte chemoattractant protein 1
- MLC
- myosin light chain
- MMP
- matrix metalloproteinase
- Palb
- glomerular permeability to albumin
- PKC
- protein kinase C
- RBF
- renal blood flow
- RPP
- renal perfusion pressure
- TGF
- transforming growth factor
- VSMC
- vascular smooth muscle cell
- WT
- wild type
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}