


Abstract
Although several antidepressants (including fluoxetine, imipramine, citalopram, venlafaxine, and duloxetine) are known to inhibit the serotonin transporter (SERT), whether or not these molecules compete with 5-hydroxytryptamine (serotonin) (5-HT) for binding to SERT has remained controversial. We have performed radioligand competition binding experiments and found that all data can be fitted via a simple competitive interaction model, using Cheng-Prusoff analysis (Biochem Pharmacol 22:3099–3108, 1973). Two different SERT-selective radioligands, [3H]N,N-dimethyl-2-(2-amino-4-cyanophenyl thio)-benzylamine (DASB) and [3H]S-citalopram, were used to probe competitive binding to recombinantly expressed human SERT or native SERT in rat cortical membranes. All the SERT inhibitors that we tested were able to inhibit [3H]DASB and [3H]S-citalopram binding in a concentration-dependent manner, with unity Hill coefficient. In accordance with the Cheng-Prusoff relationship for a competitive interaction, we observed that test compound concentrations associated with 50% maximal inhibition of radiotracer binding (IC50) increased linearly with increasing radioligand concentration for all ligands: 5-HT, S-citalopram, R-citalopram, paroxetine, clomipramine, fluvoxamine, imipramine venlafaxine, duloxetine, indatraline, cocaine, and 2-β-carboxy-3-β-(4-iodophenyl)tropane. The equilibrium dissociation constant of 5-HT and SERT inhibitors were also derived using Scatchard analysis of the data set, and they were found to be comparable with the data obtained using the Cheng-Prusoff relationship. Our studies establish a reference framework that will contribute to ongoing efforts to understand ligand binding modes at SERT by demonstrating that 5-HT and the SERT inhibitors tested bind to the serotonin transporter in a competitive manner.
SERT mediates rapid clearance of synaptic and circulating 5-hydroxytryptamine (serotonin) (5-HT), thereby playing a critical role in the spatiotemporal fine-tuning of 5-HT signaling. Multiple classes of antidepressants, including tricyclics (e.g., imipramine and clomipramine), serotonin-selective reuptake inhibitors (e.g., fluoxetine and paroxetine), and serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and venlafaxine) target SERT (Owens et al., 1997; Tatsumi et al., 1997). This transporter is also a target for drugs of abuse, including cocaine and amphetamine (Amara and Sonders, 1998). Although it is well established that these antidepressants and cocaine inhibit SERT, a clear understanding of the mode of binding to SERT has been lacking. Based on inhibition of 5-HT uptake or radioligand binding studies, different modes, including competitive, noncompetitive, and allosteric inhibition, have been proposed (Talvenheimo et al., 1979; Wennogle and Meyerson, 1982; Plenge and Mellerup, 1985; Thomas et al., 1987; Sur et al., 1998; Hummerich et al., 2004; Chen et al., 2005a). For example, conflicting data exist suggesting that imipramine competitively inhibits 5-HT uptake into platelets (Talvenheimo et al., 1979) but is allosterically influenced by 5-HT in its dissociation kinetics (Wennogle and Meyerson, 1982).
SERT belongs to the solute carrier 6 (SLC6A) Na+/Cl--coupled transporters. This gene family also includes the closely related norepinephrine transporters (NET) and dopamine transporters. The secondary structure of the SERT polypeptide is predicted to contain 12 transmembrane (TM) spanning domains with amino and carboxyl termini within the cytoplasmic compartment (Ramamoorthy et al., 1993). The three-dimensional structure of SERT is not known; however, predictive models have been built based on the crystallographic structure of the bacterial leucine transporter LeuT (Yamashita et al., 2005; Jørgensen et al., 2007a,b). This LeuT-based SERT model seems to be in agreement with mutational analysis focusing on SERT substrate and inhibitor affinities, as well as with experimental delineation of the aqueous pore in SERT using substituted cysteine accessibility studies (Chen et al., 1997; Barker et al., 1999; Chen and Rudnick, 2000; Larsen et al., 2004; Mitchell et al., 2004; Sato et al., 2004; Henry et al., 2006; Zhang and Rudnick, 2006; Plenge et al., 2007). These studies suggest an overlap between the SERT binding site for 5-HT and a site(s) for a limited number of inhibitors. Conversely, studies have reported more than one 5-HT binding site, and, in some cases, more than one inhibitor binding site (Plenge and Mellerup, 1985; D'Amato et al., 1987; Sur et al., 1998; Chen et al., 2005a,b; Plenge and Wiborg, 2005; Plenge et al., 2007). The high-affinity site is believed to be the one that underlies the pharmacological and clinical effects of antidepressants amphetamine and cocaine. For S-citalopram, the high-affinity site (site-1; Kd = 1 nM) is believed to attenuate 5-HT uptake through the transporter, whereas the low-affinity site (Kd = 3 μM) apparently modulates ligand affinity at site-1 in an allosteric manner (Plenge and Mellerup, 1985; Schloss and Betz, 1995; Plenge and Mellerup, 1997; Chen and Justice, 1998; Sur et al., 1998; Chen et al., 2005a,b; Plenge and Wiborg, 2005). Binding kinetics studies using radiolabeled citalopram, paroxetine, and imipramine has revealed that SERT inhibitors, including citalopram, exert allosteric effects at concentrations that are not relevant to their actions at the high-affinity binding site (Plenge and Mellerup, 1985; Schloss and Betz, 1995; Chen and Justice, 1998; Sur et al., 1998). Furthermore, recent crystallographic models of LeuT in complex with SERT inhibitors clomipramine, imipramine, and desipramine indicate that these compounds at high concentrations bind to a nonoverlapping site relative to the LeuT substrate, leucine (Singh et al., 2007; Zhou et al., 2007). Those inhibitors were mapped to a region of LeuT located near the extracellular gate, on the extracellular side of the substrate binding pocket (Singh et al., 2007; Zhou et al., 2007), revealing potential binding of SERT inhibitors to multiple sites at SERT.
We have examined the hypothesis that 5-HT and SERT inhibitors bind competitively to the high-affinity site (site-1) at SERT. We applied analyses based on both the Cheng-Prusoff equation (Cheng and Prusoff, 1973) and a variant of the Scatchard analysis (Jacobs et al., 2007) and tested for competitive, mutually exclusive binding of 5-HT and SERT inhibitors using high-affinity SERT-selective radioligands [3H]N,N-dimethyl-2-(2-amino-4-cyanophenyl thio)-benzylamine (DASB) and [3H]S-citalopram (Hummerich et al., 2004; Plenge and Wiborg, 2005). Based on these analyses, we provide strong evidence that several inhibitor chemotypes compete with each other and with 5-HT for binding to the high-affinity (site-1) present in the wild-type serotonin transporter.
Materials and Methods
Materials. Human embryonic kidney-293 cells stably expressing the human serotonin transporter (hSERT) (hSERT-HEK293) were kindly provided by Dr. Randy Blakely (Vanderbilt University, Nashville TN) (Qian et al., 1997). Male Wistar rats (200–250 g) were obtained from Charles River Laboratories, Inc. (Wilmington, MA). [3H]DASB (∼86 Ci/mmol) and enantiomerically pure [3H]S-citalopram (∼72 Ci/mmol) were synthesized by the Radiochemistry Group at Roche Pharmaceuticals (Palo Alto, CA). S-Citalopram, clomipramine, cocaine, duloxetine, fluvoxamine, 5-HT, imipramine, paroxetine, and venlafaxine were purchased from Sigma-Aldrich (St. Louis, MO). Indatraline was obtained from Tocris Bioscience (Ellisville, MO), and 2-β-carboxy-3-β-(4-iodophenyl)tropane (β-CIT) was obtained from Advanced Biochemical Compounds (Radeberg, Germany). R-Citalopram was purchased from Synfine Research (Richmond Hill, ON, Canada). Complete protease inhibitor cocktail was obtained from Roche Diagnostics (Mannheim, Germany). MicroScint-20 scintillation cocktail and GF/B filter plates were acquired from PerkinElmer Life and Analytical Sciences (Boston, MA).
Preparation of Membranes from hSERT-HEK293 Cells. hSERT-HEK293 cells were grown in Dulbecco's modified medium containing 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 250 μg/ml G418 (Invitrogen, Carlsbad, CA). Cells were grown to 80% confluence, harvested, and washed in calcium-free phosphate-buffered saline. For the preparation of membrane fractions, harvested cell pellets were disrupted in ice-cold 15 mM HEPES, pH 7.4, containing Complete protease inhibitor cocktail (Roche Molecular Systems, Inc., Alameda, CA), using a Polytron homogenizer (Kinematica, Littau-Lucerne, Switzerland) for 20 s at 18,000 rpm. Homogenates were centrifuged at 1000g for 15 min at 4°C. Supernatants were collected and centrifuged at 30,000g for 30 min, also at 4°C. The resulting membrane pellets were resuspended in 15 mM HEPES, pH 7.4, containing Complete protease inhibitor cocktail and stored at -80°C.
Preparation of Synaptosomal Membranes from Rat Brain Cortex. Male Wistar rats (∼200 g body weight) were euthanized with CO2 in a manner approved by the Institutional Animal Care and Use Committee at Roche Pharmaceuticals. Isolated cortices were homogenized in ice-cold sucrose buffer (0.32 M sucrose and 5 mM sodium bicarbonate, pH 7.4) using a glass homogenizer with a Teflon pestle (clearance, 0.1 mm) attached to a Wheaton overhead stirrer, applying eight strokes at 2500 rpm. Homogenates were centrifuged at 1000g for 15 min at 4°C. The resulting supernatants were collected and recentrifuged at 13,000g to yield a crude synaptosomal fraction (P2). P2 fractions were further disrupted by homogenization in 5 mM HEPES-NaOH plus Complete protease inhibitor cocktail, using a Polytron homogenizer for 20 s at 18,000 rpm. Synaptic plasma membranes and other large membranes were collected by centrifugation at 15,000g for 20 min at 4°C. The membrane pellet was resuspended in 15 mM HEPES, pH 7.4, and stored at -20°C.
Binding of 5-HT and Reuptake Inhibitors to SERT: Competition Analysis. Radioligand binding studies were conducted in assay buffer (126 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.76 mM KH2PO4, pH 7.4) containing either [3H]DASB or [3H]S-citalopram, in the absence or presence of competing agents. In [3H]DASB binding experiments, reactions were initiated by addition of membrane fractions to final dilutions of 1 μg/ml for hSERT-HEK293 membranes or 80 μg/ml for rat cortex membranes. In [3H]S-citalopram binding experiments, 5 μg/ml hSERT-HEK293 membranes were used. Incubations were allowed to proceed at 22°C for 2 h with constant shaking. Reactions were terminated by rapid filtration onto 0.5% polyethyleneimine-treated GF/B filter plates (PerkinElmer Life and Analytical Sciences). Filters were washed three times with ice-cold 15 mM HEPES, pH 7.4, buffer. Radioactivity associated with the filters was quantified using MicroScint-20 scintillant and a TopCount scintillation analyzer (PerkinElmer Life and Analytical Sciences). Nonspecific binding of [3H]DASB or [3H]S-citalopram was determined via addition of 10 μM indatraline to displace the radioligands from high-affinity binding sites. All test conditions were performed in duplicate within a given experiment, and every experiment was repeated on three to five independent occasions.
Two complimentary analytical approaches were used to assess ligand competitivity at SERT. The first method is based on the Cheng-Prusoff relationship that describes binding of two ligands in a mutually exclusive manner (Cheng and Prusoff, 1973). This relationship is described by the following equation:  where [A*] represents the radioligand concentration, Kd is the equilibrium dissociation constant of A*, KB is the equilibrium dissociation constant of the test compound, and IC50 is the test compound concentration that displaces 50% of the binding of A*. From eq. 1, a linear relationship between the IC50 value and {([A*])/(Kd) + 1} is predicted for all compounds competing with the radioligand. The slope of this line is equivalent to the affinity (KB) of the test compound. For the assessment of binding site competitivity, unlabeled compounds, diluted over a broad concentration range (1,000,000-fold), were incubated with membranes and radioligands. IC50 values and Hill slopes were determined by nonlinear regression using a four-parameter logistic equation. For this analysis, IC50 values were determined for each unlabeled test compound over a range of five to eight different radioligand concentrations ([3H]DASB, 0.1–60 nM; [3H]S-citalopram, 1–100 nM). Observed IC50 values obtained in replicate experiments were plotted against {([A*])/(Kd) + 1} for all concentrations of radioligand (A*) tested in replicate experiments. Bound radioactivity is presented as fractional receptor occupancy, calculated from the following equation:
where [A*] represents the radioligand concentration, Kd is the equilibrium dissociation constant of A*, KB is the equilibrium dissociation constant of the test compound, and IC50 is the test compound concentration that displaces 50% of the binding of A*. From eq. 1, a linear relationship between the IC50 value and {([A*])/(Kd) + 1} is predicted for all compounds competing with the radioligand. The slope of this line is equivalent to the affinity (KB) of the test compound. For the assessment of binding site competitivity, unlabeled compounds, diluted over a broad concentration range (1,000,000-fold), were incubated with membranes and radioligands. IC50 values and Hill slopes were determined by nonlinear regression using a four-parameter logistic equation. For this analysis, IC50 values were determined for each unlabeled test compound over a range of five to eight different radioligand concentrations ([3H]DASB, 0.1–60 nM; [3H]S-citalopram, 1–100 nM). Observed IC50 values obtained in replicate experiments were plotted against {([A*])/(Kd) + 1} for all concentrations of radioligand (A*) tested in replicate experiments. Bound radioactivity is presented as fractional receptor occupancy, calculated from the following equation:  where the numerator represents the specifically bound radioactivity (bound minus nonspecific) observed at any given radioligand concentration, [A*]. The denominator is calculated from the maximal specific binding (total minus nonspecific) observed at the highest (saturating) concentration of radioligand tested.
where the numerator represents the specifically bound radioactivity (bound minus nonspecific) observed at any given radioligand concentration, [A*]. The denominator is calculated from the maximal specific binding (total minus nonspecific) observed at the highest (saturating) concentration of radioligand tested.
As a validation of this method, the experimental data set was subjected to a second method of competitivity assessment based on Scatchard analyses. Bound radioactivity in the presence and absence of two fixed concentrations (10-fold apart) of each test compound was used to generate Scatchard plots. The apparent Kd for [3H]DASB and [3H]S-citalopram binding was determined for each test compound at two concentrations, one log unit apart. The Ki of the inhibitor was calculated from the increase in the apparent Kd of the radioligand using the following equation:  in which the concentration ratio was the ratio of apparent Kd of [3H]DASB or [3H]S-citalopram binding derived from Scatchard plots in the presence and the absence of the inhibitor.
in which the concentration ratio was the ratio of apparent Kd of [3H]DASB or [3H]S-citalopram binding derived from Scatchard plots in the presence and the absence of the inhibitor.
The Kd, Ki, and KB values indicated in the results all represent the equilibrium dissociation constant (k-1/k+1). The subscripts denote the different methods under which the values were determined, namely, saturation binding for radioligand (Kd), displacement binding for unlabeled test ligand (Ki), and slope of Cheng-Prusoff plot (KB), respectively.
Results
Saturable Interaction of [3H]DASB or [3H]S-Citalopram with SERT. Up to the highest concentration of 60 nM tested, [3H]DASB bound to hSERT-HEK293 membranes fitted to a simple bimolecular model, with unity Hill slope and Kd of 0.14 ± 0.04 nM (Fig. 1, A and B). For [3H]S-citalopram, up to concentrations of 100 nM, a simple, bimolecular model, with unity Hill slope and Kd of 2.4 ± 1.06 nM (Fig. 1, C and D) was obtained. The estimated numbers of saturable binding sites (Bmax) for [3H]DASB and [3H]S-citalopram were 14.9 ± 9.6 and 16.4 ± 4.2 pmol/mg protein, respectively. Linear Scatchard plots confirmed the simple bimolecular binding modes for both radioligands (Fig. 1, B and D). In preliminary studies, at concentrations greater than 60 nM for [3H]DASB and 100 nM for [3H]S-citalopram, we observed binding of [3H]DASB and [3H]S-citalopram to a second site both in hSERT-HEK293 and parent HEK293 cell membranes (data not shown). Therefore, we chose not to exceed 0.1 to 60 nM [3H]DASB or 1 to 100 nM [3H]S-citalopram for our competition binding studies
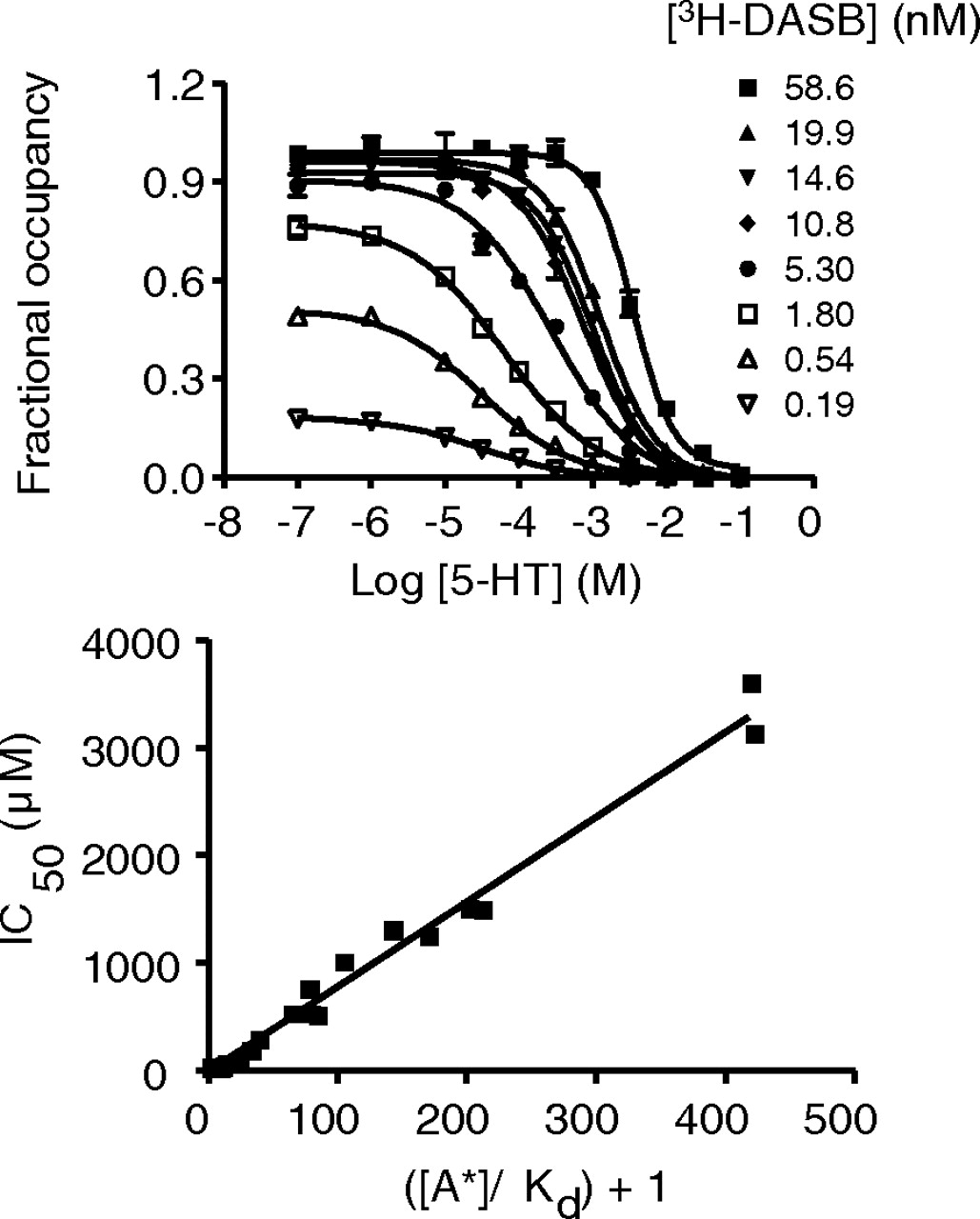
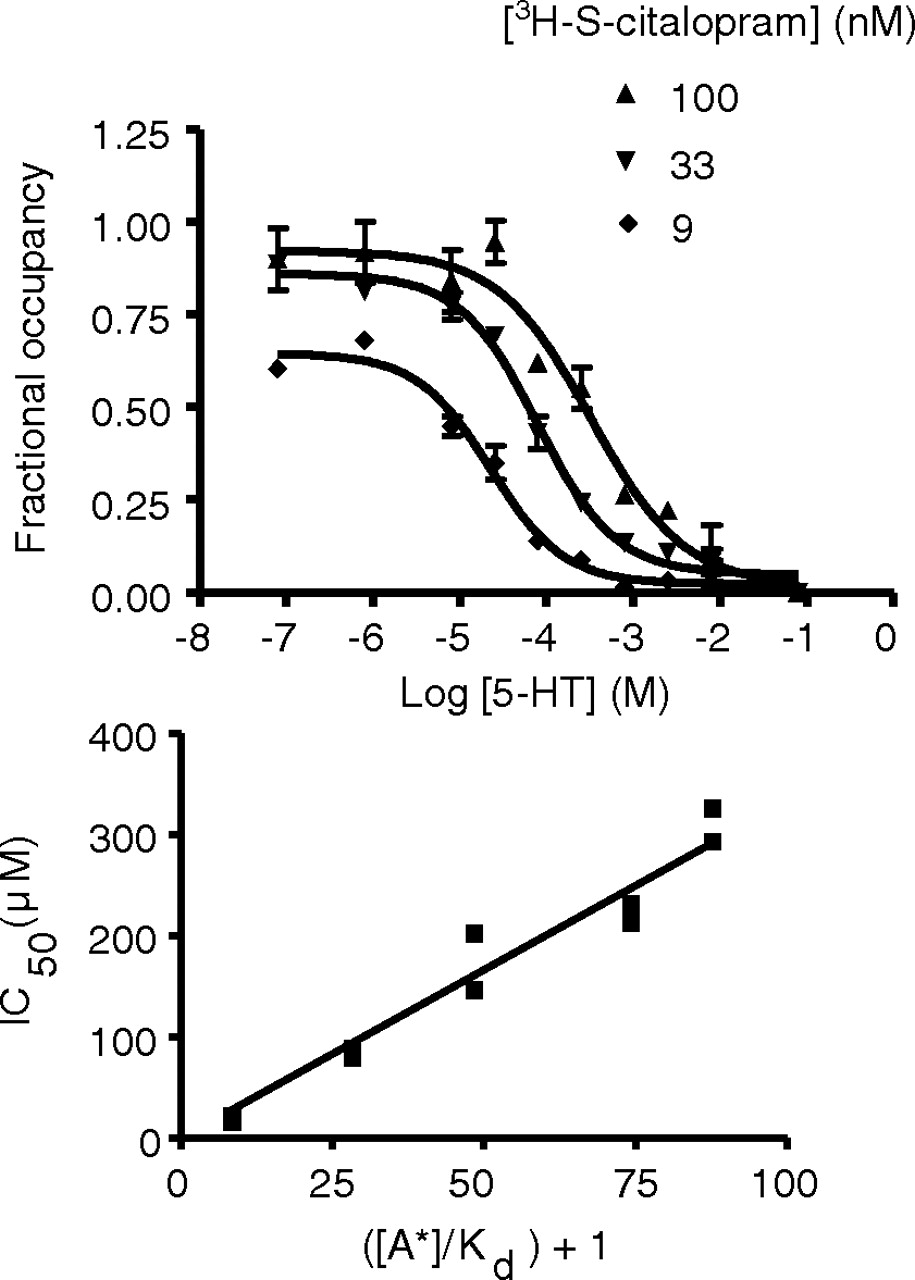
5-HT Competes with [3H]DASB and [3H]S-Citalopram for Binding at hSERT. In hSERT-HEK293 membranes, 5-HT displaced the binding of [3H]DASB or [3H]S-citalopram in a concentration-dependent manner down to nonspecific levels, as defined with 10 μM indatraline (Figs. 2 and 3). Competition between 5-HT and radioligand was explored by monitoring whether concentration-dependent displacement over a range of radioligand concentrations. In all cases, the IC50 values increased proportionally with the concentration of [3H]DASB (0.1–60 nM; Fig. 2) or [3H]S-citalopram (1–100 nM; Fig. 3). We observed linear relationships for IC50 versus {([A*])/(Kd) + 1} (r2 = 0.99 and 0.96 for [3H]DASB and [3H]S-citalopram, respectively), with estimated KB values of 7.8 ± 0.16 and 3.3 ± 0.13 μM, respectively) comparable with the previously reported Ki value for 5-HT at SERT (Bäckström et al., 1989).

Saturation binding of [3H]DASB and [3H]S-citalopram to hSERT in hSERT-HEK293 membrane fraction. Representative data showing saturation binding curves for [3H]DASB (A) and [3H]S-citalopram (C) binding to hSERT in hSERT-HEK293 membrane fraction. Nonspecific binding was determined using 10 μM indatraline. Scatchard plots of [3H]DASB (B) and [3H]S-citalopram (D) showing single-site interaction of radioligands at hSERT. The Kd and Bmax values for [3H]DASB binding to hSERT were 0.14 ± 0.03 nM and 14.9 ± 9.6 pmol/mg protein (n = 5), respectively. The Kd and Bmax values for [3H]S-citalopram binding to hSERT were 1.24 ± 1.05 and 16.4 ± 4.16 nM (n = 6), respectively.
The competitive interaction between 5-HT and DASB determined by the Cheng-Prusoff linear regression was corroborated by the Scatchard analytical method of [3H]DASB binding in the presence of 10 and 100 μM 5-HT (Fig. 4). The Bmax of [3H]DASB binding, as deduced from the x-intercept of the Scatchard plot (Fig. 4), in the presence of 10 and 100 μM 5-HT was comparable with that of control. However, the presence of 5-HT increased the apparent Kd value for DASB from 0.14 nM for control to 0.67 and 3.57 nM for 10 and 100 μM 5-HT, respectively. The calculated Ki value for 5-HT from the shifts in the apparent Kd value for [3H]DASB binding were 2.63 and 4.08 μM for 10 and 100 μM 5-HT, respectively. Similarly, the calculated Ki value for 5-HT from the shifts in the apparent Kd value for [3H]S-citalopram binding were 1.99 and 2.03 nM for 10 and 100 μM 5-HT, respectively. The average of calculated Ki values derived from [3H]DASB and [3H]S-citalopram binding by Scatchard plot analysis in the presence of 10 and 100 μM 5-HT are presented in Table 2. These Ki values were found to be comparable with the Ki values obtained using competition binding analysis (Table 1) and to the KB values (Table 2) obtained using the Cheng-Prusoff relationship based calculations.
5-HT and SERT inhibitors competitively inhibit [3H]DASB and [3H]S-citalopram binding to hSERT For the analysis using Cheng-Prusoff relationship, the IC50 values for radioligand displacement were determined for each unlabeled ligand by coincubation with multiple concentrations of radioligand in at least three separate experiments. These data were pooled to assess the relationship between observed IC50 value and (([A*]/Kd) + 1). Linear regression coefficients (r2) and equilibrium dissociation constants (KB) for 5-HT and SERT inhibitors, as determined from the observed slopes, are presented. For analysis of competitivity by the Scatchard method, the apparent Kd values for [3H]DASB and [3H]S-citalopram binding were determined from the slope of the Scatchard plots in presence of test compound at two concentrations, 1 log unit apart (see 5-HT example in Fig. 4). The Ki of the inhibitor was calculated using the equation Ki = [Inhibitor]/(concentration ratio – 1), in which the concentration ratio was calculated from the apparent Kd value of [3H]DASB or [3H]S-citalopram binding in the presence and the absence of the inhibitor. The average and the standard deviation of the two Ki values derived using the two concentrations of 5-HT or SERT inhibitors are presented. For these analyses, the equilibrium binding affinities of the radioligands, Kd values, were found to be 0.14 and 1.2 nM for [3H]DASB and [3H]S-citalopram, respectively.
Inhibition of [3H]DASB and [3H]S-citalopram binding to hSERT by 5-HT and SERT inhibitors Assay concentrations of [3H]DASB and [3H]S-citalopram were 0.5 and 1 nM, respectively. Observed IC50 values for each test compound, obtained by nonlinear regression of the concentration binding data, were converted to equilibrium binding affinities using the Cheng-Prusoff equation, under the assumption that the binding interactions were competitive. Hill slopes and Ki values obtained for different inhibitors (n = 2–4) are presented as mean ± standard deviation. For these analyses, the equilibrium binding affinities of the radioligands, Kd values, were determined from separate analyses (see Fig. 1) and were found to be 0.14 and 1.2 nM, respectively, for [3H]DASB and [3H]S-citalopram.
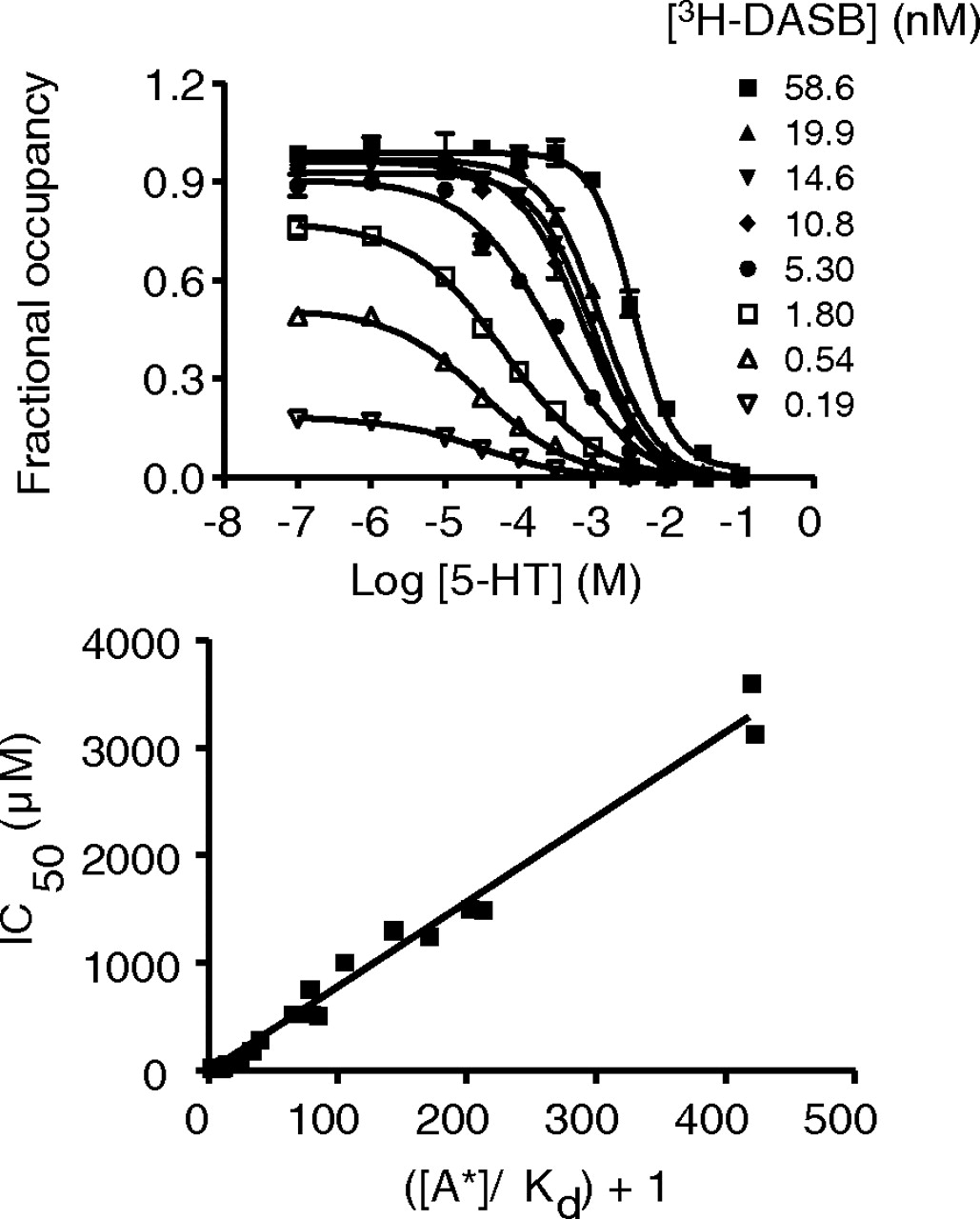
Competitive inhibition of [3H]DASB binding to hSERT by 5-HT. Representative curves (top) showing 5-HT inhibition of [3H]DASB binding to hSERT in hSERT-HEK293 membrane fraction. Linear relationship (bottom) between ([A*]/Kd) + 1 and IC50 for 5-HT showing competitive inhibition of [3H]DASB binding by 5-HT. Data from three experiments were pooled to generate the linearity plots. The Kd value for [3H]DASB used was 0.14 nM, and the estimated KB value for 5-HT was 7.8 ± 0.16 μM.

Competitive inhibition of [3H]S-citalopram binding to hSERT by 5-HT. Representative curves (top) showing 5-HT inhibition of [3H]S-citalopram binding to hSERT in hSERT-HEK293 membrane fraction. Linear plot (bottom) between ([A*]/Kd) + 1 and IC50 for 5-HT showing competitive inhibition of [3H]S-citalopram binding to hSERT in hSERT-HEK293 membrane fraction. Data were obtained from composite data generated from three independent experiments. The Kd value for [3H]S-citalopram was 1.2 nM, and the estimated KB value for 5-HT was 3.3 ± 0.13 μM.
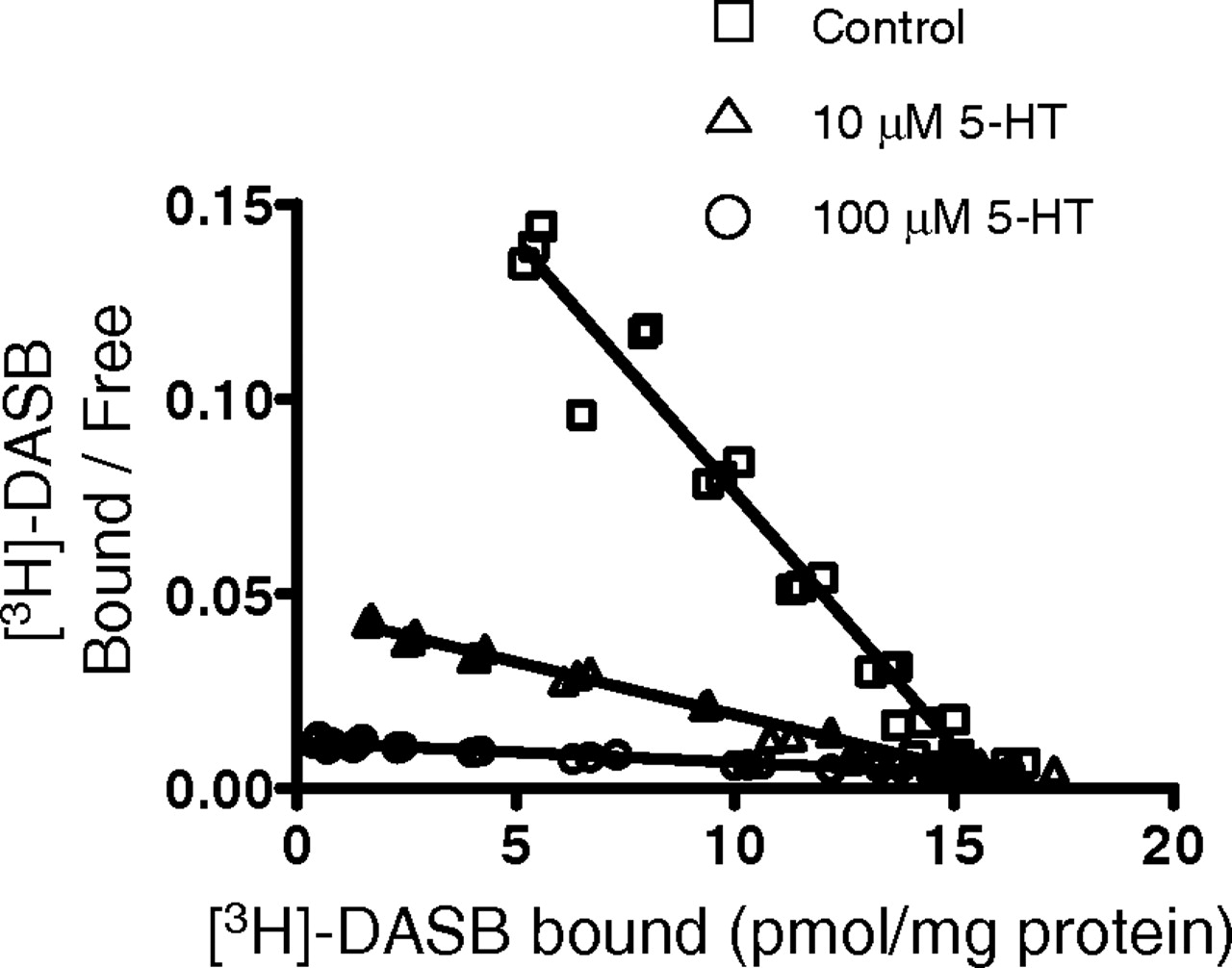
Competitive inhibition of [3H]DASB binding to hSERT by 5-HT. Scatchard plots of [3H]DASB binding to hSERT in hSERT-HEK293 membrane fraction are shown in the presence of 0, 10, and 100 μM 5-HT (n = 1). The estimated Bmax (picomoles per milligram of protein) for [3H]DASB binding to hSERT were 13.9, 14.3, and 14.0 for control, 10, and 100 μM 5-HT, respectively. The apparent Kd values for [3H]DASB binding to hSERT were 0.14 nM, 0.67 and 3.57 nM for control, 10 and 100 μM 5-HT, respectively. The Ki value of 5-HT derived using the shifts in the apparent Kd value of [3H]DASB binding were 2.63 and 4.08 μM, respectively.
Reuptake Inhibitors Compete with [3H]DASB and [3H]S-Citalopram for Binding at hSERT. In hSERT-HEK293 membranes, binding of both [3H]DASB and [3H]S-citalopram to SERT was displaced by several reuptake inhibitors in a concentration-dependent manner (Table 1). The affinity estimates of most SERT inhibitors were similar to values published previously and were associated with unity Hill slopes (Table 1) as reported previously (Owens et al., 1997; Tatsumi et al., 1997). However, the Ki and KB values for DASB and duloxetine were found to be lower than the reported values for DASB (Wilson et al., 2002) and duloxetine (Bymaster et al., 2001). In preliminary studies, we found the Ki value for certain high-affinity SERT inhibitors (e.g., duloxetine and paroxetine) to be sensitive to the concentration of transporter used in the assay. Increasing receptor concentration increased ligand depletion, thereby underestimating ligand affinity and yielding higher Ki values (data not shown). The differences in the observed Ki or KB values compared with previous reports may be due to differences in ligand-transporter equilibrium and ligand depletion.
As was the case for 5-HT, all of the reuptake inhibitors that we tested, including S-citalopram, R-citalopram, imipramine, clomipramine, indatraline, β-CIT, duloxetine, paroxetine, venlafaxine, fluvoxamine, and cocaine were found to compete with [3H]DASB or [3H]S-citalopram for interaction with SERT. Specifically, all compounds inhibited [3H]DASB binding in a concentration-dependent manner, down to nonspecific levels. For each compound, the IC50 value increased proportionally to the radioligand concentration, yielding a linear plot for the Cheng-Prusoff relationship, with r2 values ranging from 0.78 to 0.99 (Table 2). In addition, both DASB and S-citalopram were found to be competitive with each other. This is evident from the linear increases in IC50 value with radioligand concentration observed with 1) DASB versus [3H]S-citalopram and 2) S-citalopram versus [3H]DASB. KB estimates for all test compounds from the slopes of IC50 versus {([A*])/(Kd) + 1} (Table 2) were similar to affinities determined in a separate series of competition binding studies (Ki; Table 1).
To validate the results using the Cheng-Prusoff relationship analysis, as in the case of 5-HT (Fig. 4), [3H]DASB and [3H]S-citalopram binding data obtained in the presence of two concentrations of each test compound were also subjected to Scatchard analysis, apparent Kd values for radioligands were calculated, and Ki values were determined for each concentration of the two concentrations of test compounds. The average Ki values of SERT inhibitors calculated from shifts in the apparent Kd values for [3H]DASB and [3H]S-citalopram binding are presented in Table 2. These Ki values are comparable with the KB values (Table 2) obtained using the Cheng-Prusoff relationship and to the Ki values derived from competition binding studies (Table 1).
5-HT Inhibits [3H]DASB Binding in Rat Cortical Membranes. In rat cortical membranes, saturable, displaceable binding of [3H]DASB was observed, with Kd = 0.12 ± 0.02 nM (similar to that observed at hSERT-HEK293), unity Hill slope (1.04 ± 0.03), and a linear Scatchard plot, indicating simple bimolecular interaction with SERT (Fig. 5, A and B). 5-HT and several SERT inhibitors, including S-citalopram, indatraline, and DASB, inhibited binding of [3H]DASB to rat cortical membranes, in a concentration-dependent manner. IC50 values followed the same rank order of affinities reported previously for this interaction (Owens et al., 1997; Tatsumi et al., 1997). The estimated Ki values for DASB, S-citalopram, indatraline, and 5-HT were 0.14 ± 0.06, 0.4 ± 0.11, 0.70 ± 0.61, and 2410 ± 164 nM, respectively. Additionally, the observed Hill slopes were not different from unity and, in all cases, radioligand binding was displaced down to nonspecific levels observed with 10 μM indatraline (Fig. 5C).
Using the same experimental and analytical approaches described above, we determined that 5-HT binds competitively with [3H]DASB in native SERT present in rat cortical membranes. Specifically, IC50 values for 5-HT displacement of [3H]DASB binding increased linearly versus {([A*])/(Kd) + 1}, with linear coefficient of 0.99. From the slope, KB was estimated to be 6.4 ± 0.37 μM (Fig. 5D).
Discussion
The primary conclusion of this study is that serotonin and the SERT inhibitors interact with wild-type SERT in a competitive manner with the radioligands, and by inference with each other. We base this conclusion on the linear increases in IC50 observed with increasing concentration of radioligand, which exemplifies the expectations of reversible, competitive interaction from which the Cheng-Prusoff relation was derived. Our findings of Cheng-Prusoff analysis were further corroborated by parallel Scatchard analyses of the radioligand binding data.
We used for these studies the highly selective and potent radioligands [3H]DASB and [3H]S-citalopram (Hummerich et al., 2004; Plenge and Wiborg, 2005), at multiple concentrations that exceed their equilibrium binding affinities by up to 400- or 60-fold, respectively. The high selectivity of [3H]DASB for SERT (Hummerich et al., 2004), combined with the deliberate use of the enantiomerically pure [3H]S-citalopram, allowed for careful assessment of competitive behavior over a very wide concentration range, in which both radioligands were determined to interact with SERT following a seemingly bimolecular reaction. Importantly, this study assessed binding interactions at wild-type serotonin transporters, including recombinantly expressed full-length human SERT and native SERT present in rat cortical membranes. This eliminates the possibility of confounds arising from functional effects of engineered mutations. The estimated KB values for 5-HT and all SERT inhibitors tested, determined from the slopes of the IC50 values versus {([A*])/(Kd) + 1} linear plots and Ki determinations using Scatchard analysis (Table 2) and independent displacement experiments (Table 1), were similar to those in previous reports (Owens et al., 1997; Tatsumi et al., 1997). The lower Ki or KB observed for DASB and duloxetine compared with previous reports (Bymaster et al., 2001; Wilson et al., 2002) may be due to differences in assay conditions, including incubation times and relative concentrations of ligand and receptor (ligand depletion) in these studies.
Our analysis points to the competitive binding of 5-HT and SERT inhibitors at the high-affinity (site-1) of SERT, and they are consistent with previous reports of competitive inhibition of 5-HT uptake by imipramine and paroxetine (Talvenheimo et al., 1979; Thomas et al., 1987). In contrast, DASB was found to produce noncompetitive inhibition of 5-HT transport (Hummerich et al., 2004). Although the latter observation implies that DASB and 5-HT recognize different binding sites or different transporter conformations, it may equally be explained by a failure to meet steady-state conditions in those experiments.
Based on the well known alternating access model for transport function (Rudnick, 2006), it is believed that plasma membrane transporters such as SERT function by alternately exposing the substrate binding site to the extracellular (outward) and cytoplasmic (inward) aspects of the plasma membrane. Thus, the transport reaction is suggested to involve at the least in the following steps: 1) binding of the substrate to the outward-facing state, 2) isomerization of the transporter-substrate complex to the inward facing state, and 3) substrate dissociation from the inward-facing state. The alternating access model predicts the binding of 5-HT to the outward- and inward-facing transporter states (Rudnick, 2006). Additional conformations proposed in electrophysiological studies on SERT and crystallographic studies on LeuT suggest the existence of open-open and closed-closed transporter states (Adams and DeFelice, 2002; Yamashita et al., 2005; Singh et al., 2007). Our finding that reuptake inhibitors and 5-HT competitively interact for binding to SERT suggests that antidepressants and cocaine produce inhibition of transport function by blocking the initial event of 5-HT binding to the transporter. However, the states of the transporter that are sensitive to antidepressant binding remain unclear. Cysteine accessibility mutagenesis studies suggest that cocaine may stabilize the outward-facing state of SERT (Zhang and Rudnick, 2006). Conversely, ibogaine, a hallucinogenic alkaloid, has been shown to produce a noncompetitive inhibition of SERT function, presumably by stabilizing the inward facing state of SERT (Jacobs et al., 2007). Similar accessibility studies on antidepressants are lacking, but they may provide insight into the transporter states that are affected by different antidepressants.

5-HT competitively inhibits [3H]DASB binding to rat cortex synaptosomal membrane. A, representative binding data showing saturation binding of [3H]DASB to rat cortex synaptosomal membrane. Nonspecific binding was determined using 10 μM indatraline. The Kd and Bmax for [3H]DASB binding were 0.12 ± 0.02 nM and 0.42 ± 0.023 pmol/mg protein (n = 5), respectively. B, Scatchard plot of [3H]DASB binding showing single-site binding for [3H]DASB in rat cortex synaptosomal membrane. C, concentration-dependent inhibition of 0.5 nM [3H]DASB binding by indatraline, DASB, S-citalopram, and 5-HT. The estimated Ki values for DASB, S-citalopram, indatraline, and 5-HT were 0.14 ± 0.06, 0.4 ± 0.11, 0.70 ± 0.61, and 2410 ± 164 nM, respectively. D, linear relationship between ([A*]/Kd) + 1 and IC50 for 5-HT showing competitive inhibition of [3H]DASB binding in rat cortex synaptosomal membrane fraction. Data from two separate experiments were pooled to generate the linearity plots (r2 = 0.99). The Kd value for [3H]DASB binding was 0.12 nM, and the estimated KB value for 5-HT was 6.4 ± 0.37 μM.
To date, the binding sites of 5-HT and antidepressants on SERT are poorly described. The binding experiments in this study examine single events or summation of multiple-step interactions involved in the translocation of the substrate by the transporter, and they do not allow the evaluation of multiple potential modes of interaction among inhibitors, substrate, and transporter during the substrate transport cycle. Our data cannot solve which, if any, of the 5-HT binding sites also interact with inhibitor(s); we can only deduce that inhibitor and 5-HT bind in a mutually exclusive but non-necessarily syntopic manner. Recent homology modeling based on the crystal structure of LeuT, as well as cysteine accessibility mutagenesis studies are beginning to provide insight into binding pockets important for substrate and inhibitor interactions (Yamashita et al., 2005; Rudnick, 2006; Zhang and Rudnick, 2006; Jørgensen et al., 2007a,b; Singh et al., 2007; Zhou et al., 2007). The reported LeuT structure point to the presence of a tapered water-filled pore lined by hydrophilic amino acid residues located within transmembrane domains 1, 3, 6, and 8 (Jørgensen et al., 2007a,b). One binding site of 5-HT is proposed to be in the deepest region of this aqueous pore, analogous to the observed binding of leucine in LeuT. Mutational analyses have established that the SERT residues Asp98 and Ile172 are critically important for 5-HT translocation as well as inhibitor binding (Chen et al., 1997; Barker et al., 1999; Larsen et al., 2004; Henry et al., 2006). Recent homology modeling studies proposes that 5-HT occupies two subpockets within the aqueous pore defined by 1) a hydrogen bond/salt bridge between the protonated amine of 5-HT and residue Asp98 (TM1) and (2) hydrophobic interactions between the hydrophobic indole ring of 5-HT and residue Ile172 (TM3) (Jørgensen et al., 2007a,b). The three structural features of citalopram—the dimethylaminopropyl chain, the fluorophenyl group, and the cyanophthalane skeleton—were proposed to occupy separate subpockets in SERT. The subpockets occupied by the ethylamine side chain and the indole group of 5-HT were occupied by the dimethylaminopropyl and cyanophthalane groups of citalopram, suggesting the presence of a topologically overlapping primary binding pocket for 5-HT and inhibitor at hSERT. Similarly, a homology model of NET suggests that the primary site for antidepressant binding overlaps with the substrate binding site (Paczkowski et al., 2007).
Surprisingly, two residues that are located within the proposed binding site for 5-HT, Tyr95 and Ile172, are important determinants of antidepressant potency, yet they are irrelevant to 5-HT transport (Henry et al., 2006). The affinity for several reuptake inhibitors of SERT variants containing the substitutions Y95F and I172M is reduced by as much as 1000-fold in single mutants, and by 10,000-fold in double mutants, but there were no changes in 5-HT transport kinetics. Additionally, cysteine mutagenesis studies have revealed that amino acid residues Ile172 and Tyr176 are protected by 5-HT or inhibitor binding, whereas Ser404 was protected by 5-HT, but not by cocaine (Chen et al., 1997; Chen and Rudnick, 2000; Mitchell et al., 2004). These studies not only suggest the involvement of distinct amino acid residues in the binding of inhibitor and substrate binding but also the involvement of distinct amino acid residues in binding and transport steps. Given that clinically relevant SERT blockers are without exception larger and more lipophilic than 5-HT, it is not surprising to find amino acid sequences that distinguish between inhibitor and substrate binding and transport. Furthermore, such findings are not inconsistent with the overlapping binding sites proposed above.
Recent crystallographic structures of LeuT in complex with the monoamine transporter inhibitors clomipramine, desipramine, and imipramine suggest that these antidepressants bind to a nonoverlapping binding pocket localized above the leucine binding site (Singh et al., 2007; Zhou et al., 2007). However, because the amino acid sequence homology between LeuT and the solute carrier 6 family of neurotransmitter reuptake transporters, including the SERT, NET, and dopamine transporter, is low (20–25%) and the binding affinities of SERT inhibitors at LeuT are ∼1 million-fold lower (Singh et al., 2007; Zhou et al., 2007), it is possible that the antidepressant binding site identified via this crystallographic approach does not accurately represent the antidepressant binding site present in monoamine transporters.
A recent study dissecting the equilibrium thermodynamics and kinetics of reuptake inhibitors binding to SERT focuses on the impact of ligand structure on binding energetics (Martin et al., 2008). Specifically, that study shows that enthalpy changes associated with ligand binding to SERT increase in proportion with the ligand polar surface area. That finding seems to confirm that the binding pocket for 5-HT and inhibitors must be substantially exposed to the bulk solvent, perhaps the aqueous pore involved in translocation. One exception to this model is the binding mode that was observed for fluvoxamine, a compound displaying a very flexible structure. Rather than being dominated by enthalpy, the binding of fluvoxamine to SERT seemed to be entropically driven. This result, taken together with our observation of the competitive behavior between 5-HT and the reuptake inhibitors and both [3H]DASB and [3H]S-citalopram, suggests that the 5-HT transit pore may allow for more than one set of topographical coordinates for mutually exclusive interaction with antidepressants.
Acknowledgments
We thank Robert Weikert (Department of Medicinal Chemistry, Roche Pharmaceuticals, Palo Alto, CA) and Mohammad Masjedizadeh (Department of Radiochemistry) for their synthesis of labeled and unlabeled S-citalopram and DASB. We also thank Patrick Weber for contributions.
Footnotes
-
This study was wholly funded by Roche Pharmaceuticals (Palo Alto, CA).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.108.142315.
-
ABBREVIATIONS: 5-HT, 5-hydroxytryptamine (serotonin); NET, norepinephrine transporter; TM, transmembrane; LeuT, leucine transporter; DASB, N,N-dimethyl-2-(2-amino-4-cyanophenyl thio)-benzylamine; h, human; β-CIT, 2-β-carboxy-3-β-(4-iodophenyl)tropane; SERT, serotonin transporter.
- Received June 13, 2008.
- Accepted September 4, 2008.
- The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}