


Abstract
The diterpene salvinorin A from Salvia divinorum has recently been reported to be a high-affinity and selective κ-opioid receptor agonist (Roth et al., 2002). Salvinorin A and selected derivatives were found to be potent and efficacious agonists in several measures of agonist activity using cloned human κ-opioid receptors expressed in human embryonic kidney-293 cells. Thus, salvinorin A, salvinorinyl-2-propionate, and salvinorinyl-2-heptanoate were found to be either full (salvinorin A) or partial (2-propionate, 2-heptanoate) agonists for inhibition of forskolin-stimulated cAMP production. Additional studies of agonist potency and efficacy of salvinorin A, performed by cotransfecting either the chimeric G proteins Gaq-i5 or the universal G protein Ga16 and quantification of agonist-evoked intracellular calcium mobilization, affirmed that salvinorin A was a potent and effective κ-opioid agonist. Results from structure-function studies suggested that the nature of the substituent at the 2-position of salvinorin A was critical for κ-opioid receptor binding and activation. Because issues of receptor reserve complicate estimates of agonist efficacy and potency, we also examined the agonist actions of salvinorin A by measuring potassium conductance through G protein-gated K+ channels coexpressed in Xenopus oocytes, a system in which receptor reserve is minimal. Salvinorin A was found to be a full agonist, being significantly more efficacious than (trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl] benzeneacetamide methane-sulfonate hydrate (U50488) or (trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl] benzeneacetamide methane-sulfonate hydrate (U69593) (two standard κ-opioid agonists) and similar in efficacy to dynorphin A (the naturally occurring peptide ligand for κ-opioid receptors). Salvinorin A thus represents the first known naturally occurring non-nitrogenous full agonist at κ-opioid receptors.
Salvia divinorum, a member of the Lamiaceae family, has been used by the Mazatec Indians of northeastern Oaxaca, Mexico, primarily for its psychoactive effects (Wasson, 1962, 1963) for many hundreds of years (for reviews, see Valdes et al., 1983; Sheffler and Roth, 2003). The active ingredient of S. divinorum is salvinorin A, a non-nitrogenous neoclerodane diterpene that represents the most potent naturally occurring hallucinogen known (Valdes et al., 1984; Siebert, 1994). Salvinorin A induces an intense, short-lived hallucinogenic experience qualitatively distinct from that induced by the classical hallucinogens lysergic acid diethylamide, psilocybin, and mescaline (Siebert, 1994). Both S. divinorum and salvinorin A have been used recreationally for their hallucinogenic properties (Giroud et al., 2000). Intriguingly, an anecdotal case report has suggested that S. divinorum may have antidepressant properties as well (Hanes, 2001).
Quite recently, we discovered that salvinorin A has high affinity and selectivity for the cloned κ-opioid receptor (KOR) and suggested, based on limited functional studies, that salvinorin A was a KOR agonist (Roth et al., 2002). We now present a detailed report on the agonist properties of salvinorin A and selected derivatives. We discovered that salvinorin A is an extraordinarily efficacious and potent κ-opioid agonist. We also found, based on structure-function studies, that the nature of the substituent on the 2-position of salvinorin profoundly affects functional activity. Together, these results support the hypothesis that the unique effects of salvinorin A on human perception are due to selective activation of KOR.
Materials and Methods
Materials. U50488, U69593, dynorphin A, norbinaltorphimine (nor-BNI) were obtained from Sigma-Aldrich (St. Louis, MO). [3H]Bremazocine was from PerkinElmer Life Sciences (Boston, MA).
Complementary DNA Clones and cRNA Synthesis for Oocyte Studies. The rat KOR was obtained from Dr. David Grandy (GenBank accession no. D16829). The human KOR cDNA was obtained from the Guthrie Research Foundation (GenBank accession no. NM000912) and subcloned into the eukaryotic expression vector pIRESNEO (Invitrogen, Carlsbad, CA); cDNAs for KIR3.1 (accession no. U01071) and KIR3.2 (accession no. U11859) were obtained from Drs. Cesar Lebarca and Henry Lester, respectively. The chimeric G protein Gq-i5 was obtained from Bruce Conklin (University of California, San Francisco), whereas Gα16 was obtained from the Guthrie Research Foundation; both constructs were verified by automated dsDNA sequencing (Cleveland Genomics, Inc., Cleveland, OH) before use. Plasmid templates for all constructs were linearized before cRNA synthesis, and the mMESSAGE MACHINE kit (Ambion, Austin, TX) was used to generate capped cRNA.
Cell Lines and Maintenance. A stable line expressing the human KOR (hKOR-293) was obtained by transfecting an hKOR expression vector (hKOR-pIRESNEO) into human embryonic kidney-293 cells (maintained and transfected as previously detailed; Roth et al., 2002) and selecting in 600 μg/ml G418. Surviving clones were expanded and characterized with one (hKOR-293) that expressed high levels of hKOR (ca. 1 pmol/mg) used for further studies.
Oocyte Maintenance and Injection. Healthy stage V and VI oocytes were harvested from mature anesthetized Xenopus laevis (Nasco, Ft. Atkinson, WI) and defolliculated enzymatically as described previously (Snutch, 1988). The oocytes were maintained at 18°C in standard oocyte buffer, ND96 (96 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES, pH 7.5), supplemented with 2.5 mM sodium pyruvate and 50 μg/ml gentamicin (Sigma-Aldrich). One day after harvest, cRNAs were injected (50 nl/oocyte) with a Drummond microinjector. Each oocyte was injected with 0.5 ng of KOR cRNA and 0.1 ng of KIR3.1 and KIR3.2 cRNA. Recordings were made at least 48 h after injection.
Electrophysiological Studies. An Axon Geneclamp 500 amplifier was used for standard two-electrode voltage-clamp experiments. The FETCHEX program (Axon Instruments, Foster City, CA) and recorded data traces were used for data acquisition and analysis. Oocytes were then removed from incubation medium, placed in the recording chamber containing ND96 medium, and clamped at -80 mV. Recordings were made in hK buffer (72.5 mM NaCl, 24 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES, pH 7.5). To facilitate the recording of inward K+ currents through the KIR3 channels, the normal oocyte saline buffer was modified to increase the KCl concentration to 24 mM K+. Microelectrodes were filled with 3 M KCl and had resistances of 0.4 to 2.0 MΩ.
Radioligand Binding and Functional Studies. Radioligand binding studies were performed as described previously (Roth et al., 2002) with the exception that 150 mM NaCl was added to the standard binding buffer to mimic physiological sodium concentrations. In brief, membranes (10-50 μg) were incubated together with [3H]bremazocine in a final volume of 0.5 ml with a buffer of the following composition: 50 mM Tris-HCl, 150 mM NaCl, pH 7.40 along with test agents for 90 min at room temperature. Incubations were terminated by rapid filtration and collection on GF/C glass fiber filters and washing with ice-cold binding buffer. Dried filters were put into sample vials, scintillation fluid was added, and dpm were measured by liquid scintillation spectroscopy. Measurements of the ability of KOR agonists to inhibit forksolin-stimulated adenylate cyclase activity were performed as detailed previously (Roth et al., 2002). For studies involving measurements of intracellular calcium mobilization, a Molecular Devices FLEXSTATION was used as recently detailed (Rothman et al., 2003). For these studies, hKOR were cotransfected with the chimeric G protein Gaq-i5 (Conklin et al., 1993) or the “universal” G protein Ga16 (Offermanns and Simon, 1995). Measurements of intracellular calcium mobilization and quantification of agonist efficacy and potency were performed as described in Rothman et al. (2003).
Data Analysis. EC50 values and curve fitting were determined using Nfit software (Island Products, Galveston, TX) or GraphPad Prism (GraphPad Software, Inc., San Diego, CA). Student's t test was used for comparison of independent means, with values reported as two-tailed p values.
Chemistry. Salvinorin A was isolated from dried leaves of S. divinorum by the method reported previously (Valdes et al., 1984). Salvinorin A was hydrolyzed using potassium carbonate in methanol to yield salvinorin B. The reported esters were formed using salvinorin B, dimethylaminopyridine, and the corresponding acid chloride in methylene chloride.
Salvinorin B was characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) and found to be authentic by comparison with literature values (Valdes et al., 1984). The reported esters were purified by high-performance liquid chromatography and characterized by HRMS. 1H and 13C NMR spectra were recorded on a Bruker AMX 500 MHz NMR spectrometer in CDCl3. The HRMS were measured using a Bioapex FT mass spectrometer with electrospray ionization. High-performance liquid chromatography was conducted on a Waters Deltaprep 4000 system using a Waters Xterra RP18, 5 μm, 4.6 × 150-mm column, with mobile phase H2O/acetonitrile (1:1). Thin layer chromatography analyses were carried out on precoated Si gel G254, 250-μm plates, with the developing system hexane/ethyl acetate (2:1) and visualized with vanillin/H2SO4 in ethanol.
Preparation of Esters. Salvinorin B (10 mg, 26 nmol) and 4-dimethylaminopyridine (catalytic amount) were dissolved in methylene chloride (3 ml). The corresponding acid chloride (130 nmol) was added, and the reaction stirred at room temperature overnight. The mixture was quenched with methanol, loaded onto silica, and purified by vacuum liquid chromatography using Si gel (230-400-mesh) with hexane/ethyl acetate (3:1) solvent system. Calculated molecular weights were obtained using ChemDraw software (Table 1).
Calculated molecular weights were obtained using Chem Draw software
Results
In initial studies, we examined the abilities of salvinorin A and selected derivatives (see Fig. 1 for structures) for their ability to bind to hKORs. As can be seen, the synthetic derivatives of salvinorin A differ solely in the nature of the substituent in the 2-position. As is shown in Table 2, salvinorinyl-2-propionate was the only derivative with submicromolar affinity for hKORs; also of note is that salvinorin B was inactive at hKORs. A screen of a number of other receptor subtypes showed that the salvinorin A derivatives tested had no significant activity at other receptors, including various serotonergic, dopaminergic, muscarinic, adrenergic, cannabinoid, and σ receptors (see Table 2 for details)

Structures of salvinorin A, B, and 2-salvinorinyl esters. Shown are the structures of the compounds used in this study.
Effect of salvinorin A derivatives on KOR binding and inhibition of forskolin-stimulated adenylate cyclase in KOR-293 cells
Shown are the mean values ± S.D. from n = 2-4 separate experiments in which Ki values for inhibition of [3H]bremazocine binding and EC50 and Emax values for inhibition of adenylate cyclase in KOR-293 cells were performed as detailed under Materials and Methods with the response induced by U69593 defined as 100%. The salvinorin A derivatives listed above were also screened at a large number of cloned receptors and found to have no significant activity, when tested at 10 μM at the following receptors: serotonin (5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT5a, 5-HT6, 5-HT7), dopamine (D1, D2, D3, D4, D5), muscarinic (m1, m2, m3, m4, m5), μ, δ, and ORL-1 opioid receptors, σ1, σ2, α1-adrenergic (1a, 1b, 1d), α2-adrenergic (2A, 2B, 2C) β2-adrenergic, and CB-1 cannabinoid receptors [assayed as previously detailed (Shi et al., 2003)].
We next evaluated the ability of salvinorin A and the propionate and heptanoate derivatives to activate hKORs by measuring the ability to inhibit forskolin-stimulated cAMP production using U69593 as the comparator. As shown in Table 2, salvinorin A and salvinorinyl-2-propionate were potent and full agonists compared with U69593, whereas salvinorinyl-2-heptanoate was a partial agonist.
We also evaluated the ability of U69593, dynorphin A, salvinorin A, and the propionate derivative of salvinorin A to activate hKORs using a fluorescent-microplate-reader (FLEXSTATION) wherein hKORs were cotransfected with either the chimeric G protein Gqi5 or the universal G protein Ga16 as detailed previously (Rothman et al., 2003). Figure 2 shows representative results for U69593 and salvinorin A using either Gα16 (A and B) or Gq-i5 (C and D). No responses were seen in untransfected cells or in cells transfected with hKOR alone (data not shown). Figure 2 also shows a representative dose-response study using Gq-i5 as the chimeric G protein. Because both methods seemed to yield equivalent results, further studies were performed with Gq-i5. Table 3 shows representative EC50 and Emax values for a variety of KOR agonists using Gq-i5. In these studies, salvinorin A was more potent than any other of the tested KOR agonists (Table 3). In terms of maximal response, all of the active compounds gave similar responses.
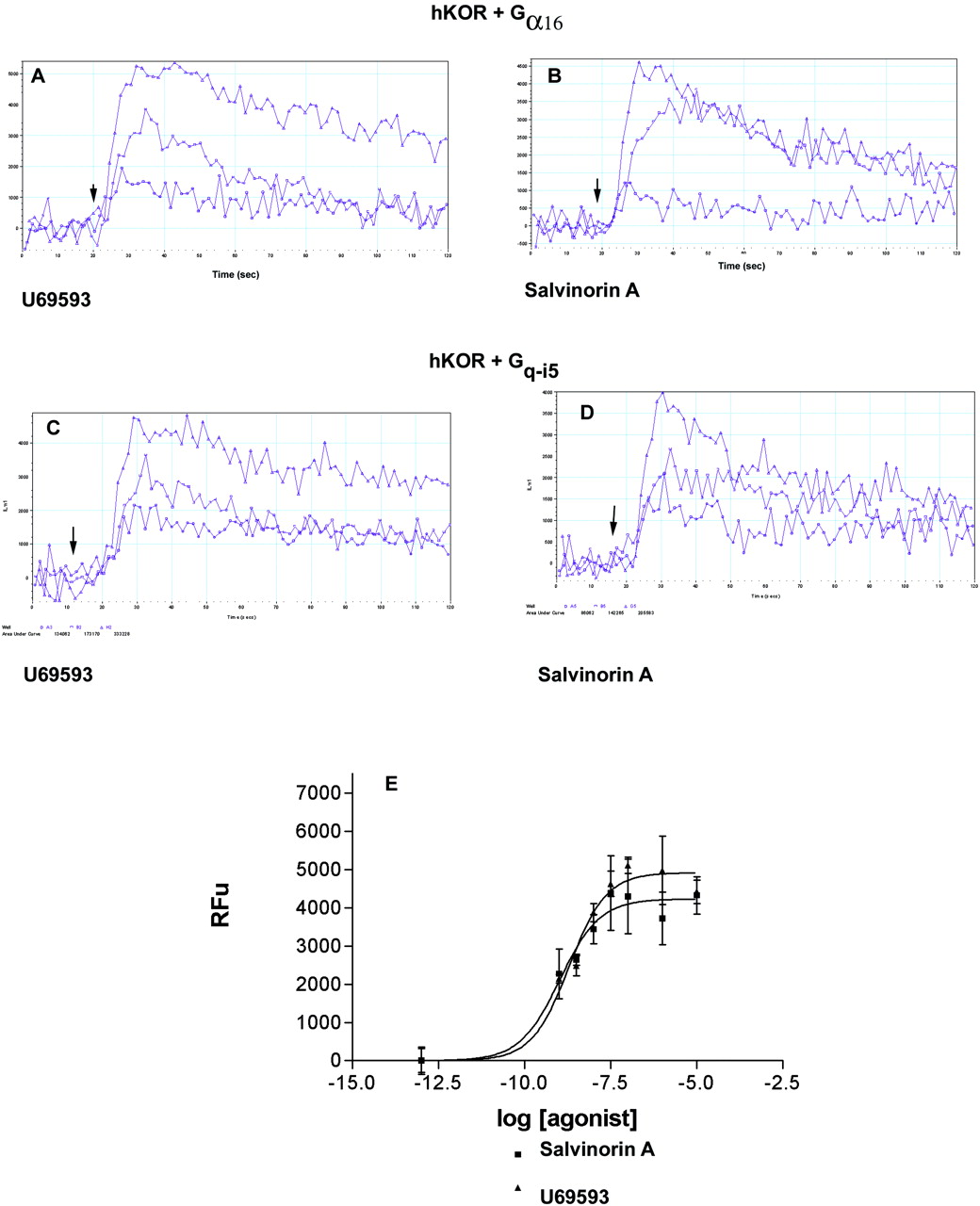
Salvinorin A mobilizes intracellular Ca2+ when hKORs are cotransfected with the universal G protein G16 or the chimeric G protein. For these studies human embryonic kidney-293 cells were transfected with hKOR and either Gqi5 or G16 and the mobilization of intracellular calcium quantified as described previously (Rothman et al., 2004) using a 96-well FLEXSTATION. A and B, representative results with increasing doses of U69593 or salvinorin A (0, 10, and 100 nM), whereas hKORs were cotransfected with G16. C and D, results obtained when hKORs were cotransfected with Gq-i5. E, average for n = 3 separate experiments for dose-response studies to salvinorin A and U69593.
Salvinorin A and salvinorinyl-2-propionate are agonist at hKOR-stimulated intracellular Ca2 mobilization: comparison with reference compounds
Data represent mean ±S.D. of quadruplicate determinations for EC50 and Emax for mobilization of intracellular calcium.
It is well known that overexpression systems tend to provide inaccurate estimates of agonist potencies and efficacies because of issues of receptor reserve (Kenakin, 2002). As well, it has been well described that unnatural expression systems wherein chimeric or “universal” G proteins are used also lead to misleading estimates of agonist potencies and maximal responses (Woolf et al., 2001; Kenakin, 2002). Accordingly, we next determined the maximal agonist responses (Emax) and potencies (EC50 values) of selected compounds using a system without receptor reserve.
Salvinorin A Is a Full agonist. For these studies, Xenopus oocytes were coinjected with inwardly rectifying K+ channels and KORs. In the experiment shown, a representative oocyte voltage clamped at -80 mV was first perfused with hK buffer (containing 24 mM KCl) to shift the reversal potential of potassium and facilitate K+ current through Kir3 (Fig. 3). Perfusion with 1 μM salvinorin A significantly increased the inward current, and the activation was reversed by 100 nM nor-BNI. Similarly, 1 μM U69593 increased the inward current in a different oocyte, and the effect was also blocked by 100 nM nor-BNI (Fig. 1B). Neither 10 μM salvinorin A nor U69593 increased the membrane conductance of oocytes expressing Kir3 without KOR (data not shown).

Salvinorin A is a highly efficacious κ-receptor agonist. Representative traces showing the change in current during a typical experiment. A large inward current was apparent as the K+ concentration was increased from 2 to 24 mM in normal oocyte saline buffer. Salvinorin A (1 μM) and U69593 (1 μM) in the buffer (24 mM K+) further increased Kir3 currents, and the response was reversed by nor-BNI (100 nM), a κ-antagonist.
Concentration-response curves of salvinorin-A and κ-agonists U69593 and U50488 were compared (Fig. 4). Each point represents the mean response measured in four to seven different oocytes. Data were collected from multiple batches of oocytes and merged by normalizing the responses to the average maximal response produced by salvinorin A on that recording day. Based on these results, salvinorin A was not significantly more potent (EC50 = 69 nM; confidence intervals 50-94 nM) than U69593 (EC50 = 224 nM; confidence intervals 51-157 nM) or U50488 (EC50 150 nM; confidence intervals 50-194 nM).
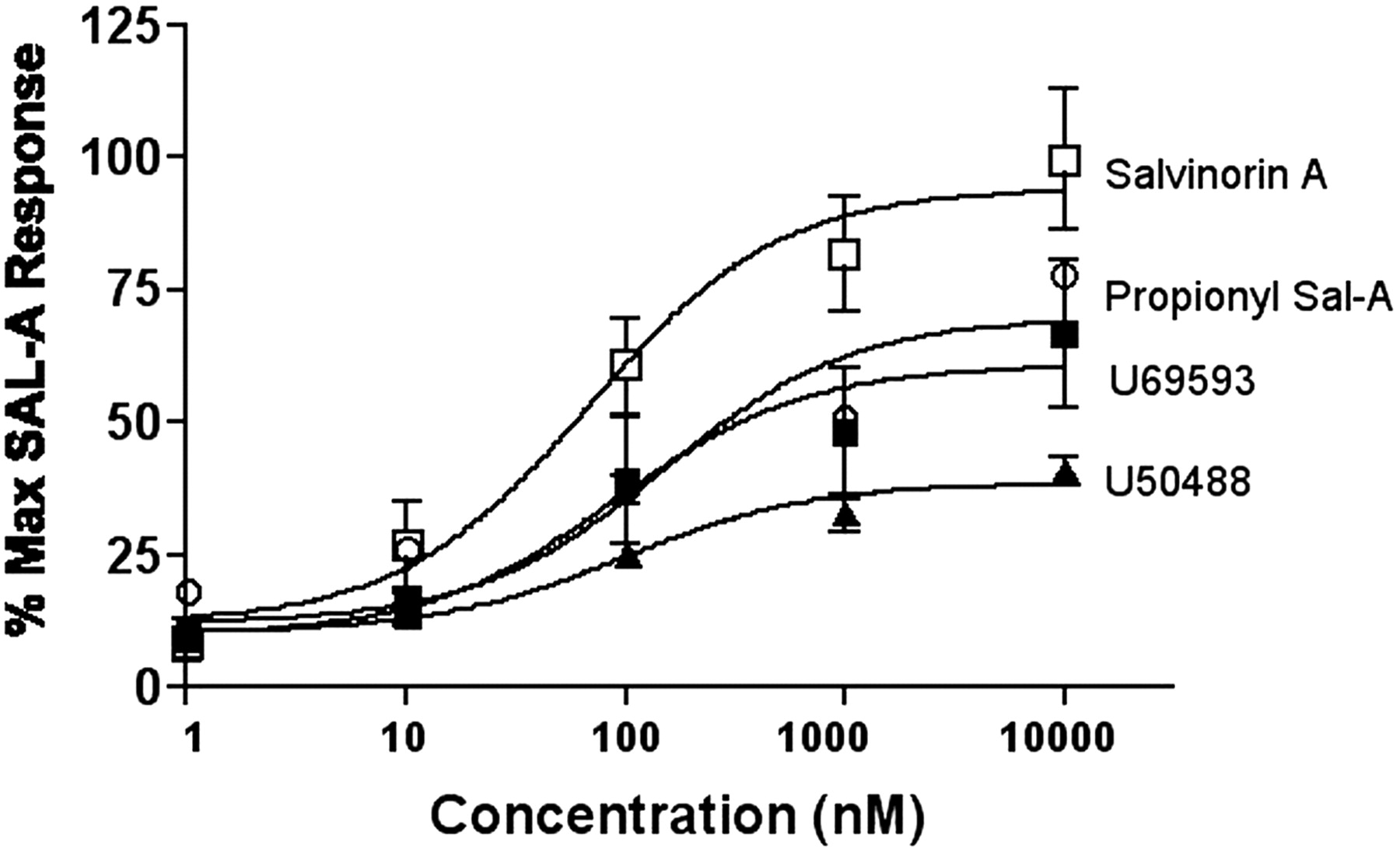
Concentration-response curve of salvinorin A, and κ-agonists U69593 and U50488. Cumulatively higher concentrations of salvinorin A and the κ-agonists were applied to the bath. The agonist response at each concentration was normalized as a percentage of the maximal salvinorin A response. Each point represents the mean response measured in four to seven different oocytes.
Under these expression conditions, there was an apparent lack of spare κ-receptors. Increasing the κ-receptor cRNA from 0.5 ng/oocte to 1.0 ng increased the average U69593 response from 1.63 ± 0.57 to 2.76 ± 1.04 μA (n = 7 or 8). Based on the lack of spare receptors, we directly compared the maximal responses evoked by 10 μM each of the κ-agonists (Fig. 5) with that of dynorphin A. In this assay, propionyl-salvinorin also acted as a partial agonist whose maximal activity was less than salvinorin A. The response to salvinorin A was significantly greater than that to U69593 and U50488 (p < 0.05), but not significantly greater to that of dynorphin A.

Salvinorin A is more efficacious than U69593 and U50488 in κ-receptor-mediated activation of Kir3 currents. At saturating concentration, salvinorin A (10 μM) evoked a large Kir3 currents, which were significantly higher than the response evoked by U69593 (10 μM) or U50488 (10 μM). Data are mean ± S.E.M.; **, p < 0.05. Dynorphin A (10 μM) produced a response that was not significantly different from salvinorin A.
Discussion
The principal finding of this study is that salvinorin A is an extraordinarily potent full agonist at hKORs. Additionally, we report that salvinorinyl-2-propionate is a potent partial agonist at KORs and also demonstrate that the nature of the 2-substituent of the salvinorin scaffold is critically important for agonist efficacy and potency. We also have obtained data with KOR-knockout and wild-type mice that the actions of salvinorin A are mediated by KOR in vivo (J. Pintar, personal communication). Together, these results imply that the profound effects of salvinorin A on human consciousness are mediated by potent and highly efficacious activation of KORs.
In prior reports, we have suggested that because salvinorin A is a potent hallucinogen that is apparently selective for KORs, and that targeting KORs might lead to novel medications for the treatment of diseases manifested by hallucinatory experiences (e.g., schizophrenia, affective disorders, and dementia) (Roth et al., 2002; Sheffler and Roth, 2003). In this regard, studies with nonselective opioid antagonists that possess KOR actions in schizophrenia have been mixed (Rapaport et al., 1993; Sernyak et al., 1998), although there are no studies in which selective KOR antagonists have been tested. Because of anecdotal reports that extracts of S. divinorum may possess antidepressant actions (Hanes, 2001), and published studies in rodents that KOR antagonists block stress-induced responses (McLaughlin et al., 2003), KOR antagonists could possess antianxiety/antidepressant actions as well. Indeed, a recent study (Mague et al., 2003) suggested that κ-selective antagonists might have intrinsic antidepressant actions. Our current studies suggest that novel KOR-selective agents might be obtained by selective modification of the salvinorin scaffold. Whether such agents might possess antidepressant or antipsychotic activity is unknown.
As shown in these studies, salvinorin A and salvinorinyl-2-propionate are potent agonists at KORs with salvinorin A being a full agonist in most assay systems, whereas salvinorinyl-2-propionate is likely a partial agonist. Salvinorin B and all other tested salvinorin derivatives were devoid of significant activity. One potential complication of the studies performed on recombinant, overexpressed receptors relates to the issue of receptor reserve. Thus, it is widely appreciated that overexpressing G proteins and/or receptors in heterologous expression systems leads to inaccurate estimates of agonist potencies and maximal responses (for review, see Kenakin, 1997). Accordingly, we also evaluated the agonist actions of salvinorin A and other compounds at KORs expressed in Xenopus oocytes.
KOR expressed in Xenopus oocytes activate intrinsic G proteins that then increase the conductance of coexpressed G protein-coupled inwardly rectifying potassium channels (GIRK and Kir3) (Henry et al., 1995). Injection of cRNAs coding for the mammalian receptor and channel has been demonstrated to faithfully reconstitute opioid signaling in oocytes equivalent to that observed in guinea pig substantia gelatinosa neurons (Grudt and Williams, 1993). In addition, by controlling the levels of receptor and channel expression, spare receptors can be avoided and the peak responses produced by different drugs can be a direct measure of agonist efficacy. The in vitro bioassay also eliminates pharmacokinetic barriers, and the electrophysiological recording of channel activation provides a rapid measure of receptor activation. In this study, we compared the relative activity of salvinorin A with three compounds having established κ-opioid receptor agonist activity. Salvinorin A was found to be more potent and have higher efficacy than either U50488 and U69593. The agonist efficacy of salvinorin A was not significantly different from dynorphin A(1-17), an endogenous neurotransmitter of the κ-opioid receptor (Chavkin et al., 1982).
Structure-activity relationship studies show that the KOR agonistic activity of salvinorin derivatives depend largely on the size and character of the substituent on the 2-ester moiety. Generally, the studied derivatives have either lower affinity for KOR than salvinorin A or are completely devoid of activity. The two active derivatives, the propionate and the heptanoate, demonstrate that as the alkyl chain is lengthened, KOR affinity diminishes. Interestingly however, chain length must not be the only factor, because the short-chain ethylcarbonate derivative is absent of activity.
The current results support the conclusion that just as morphine is a natural plant product able to activate the μ-opioid receptor, salvinorin A is a natural plant product able to activate the KOR. The strongly psychotomimetic actions of salvinorin A suggest that the dynorphin/κ-opioid system may have a role in the regulation of cognition and perception and support earlier proposals that some forms of schizophrenic hallucinations may be caused by hyperactive endogenous opioid systems (Gunne et al., 1977). Recent data implicating the KOR-dynorphinergic system in modulating stress and anxiety responses in rodents suggest that targeting KORs might also lead to novel antidepressant and anxiolytic medications. Salvinorin A, by virtue of its potency, efficacy, and selectivity as a KOR agonist will be an important tool for discovering the role that the KOR-dynorphinergic system has in health and disease.
Footnotes
-
The work was supported by U.S. Public Health Service Grant RO1 DA04123 from National Institute on Drug Abuse (to C.C.) and by the National Institute of Mental Health Psychoactive Drug Screening Program and KO2MH01366 and RO1DA017204 (to B.L.R.).
-
DOI: 10.1124/jpet.103.059394.
-
ABBREVIATIONS: KOR, κ-opioid receptor; hKOR, human κ-opioid receptor; nor-BNI, nor-binaltorphimine; U50488, (trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl] benzeneacetamide methane-sulfonate hydrate; U69593, (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide.
- Received August 31, 2003.
- Accepted November 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}