


Abstract
Inhibition of acetylcholinesterase (AChE) by certain organophosphates (OPs) can be life-threatening and requires reactivating antidote accessibility to the peripheral and central nervous systems to reverse symptoms and enhance survival parameters. In considering dosing requirements for oxime antidotes in OP exposures that inactivate AChE, clearance of proton ionizable, zwitterionic antidotes is rapid and proceeds with largely the parent antidotal compound being cleared by renal transporters. Such transporters may also control disposition between target tissues and plasma as well as overall elimination from the body. An ideal small-molecule antidote should access and be retained in primary target tissues—central nervous system (brain), skeletal muscle, and peripheral autonomic sites—for sufficient periods to reactivate AChE and prevent acute toxicity. We show here that we can markedly prolong the antidotal activity of zwitterionic antidotes by inhibiting P-glycoprotein (P-gp) transporters in the brain capillary and renal systems. We employ the P-gp inhibitor tariquidar as a reference compound and show that tissue and plasma levels of RS194B, a hydroxyl-imino acetamido alkylamine reactivator, are elevated and that plasma clearances are reduced. To examine the mechanism, identify the transporter, and establish the actions of a transport inhibitor, we compare the pharmacokinetic parameters in a P-glycoprotein knockout mouse strain and see dramatic enhancements of short-term plasma and tissue levels. Hence, repurposed transport inhibitors that are candidate or Food and Drug Administration–approved drugs, should enhance target tissue concentrations of the zwitterionic antidote through inhibition of both renal elimination and brain capillary extrusion.
SIGNIFICANCE STATEMENT We examine renal and brain capillary transporter inhibition as means for lowering dose and frequency of dosing of a blood-brain barrier permanent reactivating antidote, RS194B, an ionizable zwitterion. Through a small molecule, tariquidar, and gene knockout mice, CNS antidote concentrations are enhanced, and total body clearances are concomitantly diminished. RS194B with repurposed transport inhibitors should enhance reactivation of central and peripheral OP-inhibited acetylcholinesterase. Activities at both disposition sites are a desired features for replacing the antidote, pralidoxime, for acute OP exposure.
Introduction
Organophosphate (OP) agents, such as sarin and venomous agent X (VX), irreversibly inhibit acetylcholinesterase (AChE), causing potentially severe and irreversible damage in the central nervous system (CNS) and peripheral nervous system (PNS). A preferable strategy for treating acute OP toxicity is to administer high doses of antidote to reactivate CNS and PNS AChE. Accordingly, an ideal reactivating antidote should rapidly cross the blood-brain barrier (BBB) and maintain a high drug concentration in the CNS without being subject to rapid elimination and target tissue clearance. Many pharmacologic agents containing ionizing groups or permanent charges are subject to rapid clearance as the parent, unchanged compound (Nigam, 2015; Giacomini and Sugiyama, 2017), which may significantly decrease drug efficacy during treatment. Over the years, a plethora of transporters that control efflux from the CNS target sites and peripheral organs of the body have been identified, characterized, and studied in cell culture and intact animals (Kavas et al., 2013; Nigam 2015; Giacomini and Sugiyama, 2017). These studies have largely been designed to circumvent competitive interactions at transporters controlling tissue extrusion or overall body elimination and to recommend dosage adjustment with multiple drug administrations (Verbeeck and Musuamba, 2009; Giacomini and Sugiyama, 2017).
In fact, Joosen and colleagues (Joosen et al., 2016; Meerhoff et al., 2018) have shown that the P-glycoprotein (P-gp) inhibitor tariquidar (TQD) will inhibit atropine efflux from the multidrug resistance protein 1-Madin-Darby canine kidney MDRI-MDCK cell culture system. Taken together, these observations lead to the possibility that ionizable, zwitterionic oximes containing a basic amine are subject to disposition changes resulting from inhibition of extrusion from the CNS and elimination from the circulation by renal tubular and biliary secretion mechanisms (Kobrlova et al., 2019; Taylor et al., 2019). In turn, in combining antidotes with repurposed transport inhibitors during drug administration, concentrations in excretion fluids should be diminished, and concentrations in target tissues, the CNS and PNS, and in organs of excretion, kidney and liver, would be enhanced (Sadeque et al., 2000).
In our previous studies, a lead in a family of ionizable, zwitterionic oximes, RS194B, has been shown to be an effective reactivator in vitro of OP-conjugated and -inhibited AChE (Sit et al., 2018). Moreover, it exhibits substantial survival efficacy when administered postexposure in mice and macaques to the toxic OP, such as sarin or paraoxon (Rosenberg et al., 2017; Rosenberg et al., 2018; Sit et al., 2018). After bolus dosing, an antidote containing a nonionized species can readily cross membranes, such as the BBB, by passive diffusion (Rankovic, 2015; Gupta et al., 2019).
Even if the fraction of neutral species is in minor abundance, the surface area of the capillaries to the brain is sufficient to allow rapid passage into the CNS (Scheme 1). However, within the capillary endothelial cell in the CNS, the antidote is subject to reducing CNS accumulation (Fromm, 2000, 2004).

Ionization equilibria of RS194B, transition from cations to anions through a zwitterionic and a neutral form. The two pKa values for both the protonated amine and oxime are found around 8.8. The neutral form facilitates crossing through the BBB, and the cation and zwitterionic forms in tissues will enter the active center gorge and reactivate the AChE enzyme activity inhibited by the OP.
Accordingly, a competing P-gp inhibitor of low toxicity may slow overall clearance by the renal tubules, causing retention of the reactivating agent in the plasma. Moreover, the inhibitor may also be functional in brain capillaries to block extrusion transport from the brain and enhance CNS concentrations of the antidote (Sadeque et al., 2000).
To explore these transport and disposition issues, we have employed a reference compound, the P-gp inhibitor TQD, to reduce body clearance of the zwitterionic oxime and selectively enhance brain concentrations by inhibiting extrusion by brain capillary endothelial cells. To identify the transporter, examine the mechanism, and establish specificity, we compared the pharmacokinetic parameters in a P-gp knockout strain and see dramatic enhancements of short-term plasma and tissue levels. These findings in the mouse indicate that the P-gp and perhaps other ATP binding cassette transporters have a dominant influence on renal elimination and brain capillary extrusion.
Materials and Methods
Chemicals
TQD, heparin, DMSO, and citric acid were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Acetonitrile was from Fisher Scientific (Waltham, MA). Sterile PBS was obtained from Corning (Corning, NY), and isoflurane was purchased from VetOne (Boise, ID).
RS194B as a Lead Oxime and RS138B as a Reference Compound
RS194B was prepared as the free base as previously described (Rosenberg et al., 2017, 2018) and was formulated for intramuscular injection at a concentration of ∼70 mg/ml. RS194B was dispersed and rendered soluble by incremental dropwise addition of citric acid or dilute HCl to bring the pH to 6.5, resulting in clarity of the solution. The solution was allowed to stand for at least 24 hours at 4°C, to ensure an absence of precipitation, before injection at room temperature. The concentration of ionized species, when injected, is slightly above isotonicity. A close congener of RS194B, RS138B was dissolved in acetonitrile in a concentration of 2 μM and was used as an internal standard for RS194B during liquid chromatography/mass spectrometric (LC/MS/MS) (SCIEX, Framingham, MA) analyses (Sit et al., 2018).
Pharmacokinetics of RS194B in Mice
Mice were housed in the University of California San Diego (UCSD) vivarium. Studies were conducted in a laboratory hood, in compliance with the Animal Welfare Act and regulations stated under the guide of the Institutional Animal Care and Use Committee.
Male BALB/c mice (8 weeks of age and weighing 25–30 g), purchased from Envigo (Somerset, NJ), were administered RS194B intramuscularly into both rear thighs in divided doses of 35 mg/kg and in volumes of 50 μl (total of 70 mg/kg per mice). PBS was employed as the intramuscular injection for the control group of mice. Individual mice were bled at 5, 10, 15, 30, 60, and 120 minutes after RS194B intramuscular and intravenous injections (n = 5, at each time). Sampling was by retro-orbital bleeds. Blood samples were centrifuged immediately for plasma collection.
For organ collection and tissue analysis, mice were first perfused with PBS with trace heparin and then brain, kidney, liver, lung, diaphragm, upper limb muscle, and muscle localized to the injection site (rear thigh) were isolated and rinsed with PBS. These organs, along with urine and biliary samples from the urinary and gallbladders, were collected and analyzed similarly to the blood samples. Samples were collected 5, 10, 15, and 30 minutes after RS194B or control (PBS) intramuscular injections (n = 3). Frozen tissue samples were minced into small pieces and homogenized with tissue homogenizer for LC/MS/MS analyses.
All pharmacokinetic values are computed as follows: Cmax is extrapolation of the Cmax value back to zero time. Area under the curve (AUC) is computed by integration. The elimination rate constant is obtained from the slope of the first-order plot. Volume of distribution (Vd) represents the initial plasma concentration (or the multiple of AUC and elimination rate constant, ke) with respect to the administered dose. Clearance is evaluated from the initial drug dose and AUC.
Pharmacokinetics of RS194B in Mice with P-gp Inhibitor TQD
TQD (99.1% pure) was dissolved in 5% DMSO and sterile water, resulting in clarity of the solution. It was then injected intraperitoneally into BALB/c mice at a dose of 7.5 mg/kg in an ∼1.0-ml volume. After a 10-min interval, RS194B was administered intramuscularly, as described above. After brief anesthesia with isoflurane, blood and tissue samples were collected in separate sets of mice at 15, 30, 60, and 120 minutes after RS194B intramuscular injection (n = 3 or more samples).
Pharmacokinetics in P-Glycoprotein Knockout Strains
Multidrug resistance protein 1a/b (Mdr1a/b) mice, the P-glycoprotein constitutive knockout strain and FVB control strains), purchased from Taconic Biosciences (Rensselaer NY 12144 USA), were used to study the pharmacokinetics of RS194B, largely in the absence of P-gp transporters TQD. In all, 12 male (15, 30, 60, and 120 minutes after RS194B IM injection, n = 3) and two female (15, 30 minutes after RS194B IM injection, n = 3) P-gp knockout mice and 12 male and two female controls were used in the analysis described in the Results. Procedures for RS194B administration and body fluid and tissue collection were described above.
Tissue Homogenization and Extraction
All tissues, as ∼100-mg samples, were weighed and kept in dry ice after collection and then transferred to a −80°C freezer until homogenization and processing. Tissues were shred with a scalpel and then transferred to a clear 2-ml Eppendorf tube along with 1-mm Zirconia/Silica Beads (BioSpec, Bartlesville, OK). Acetonitrile containing 2 μM RS138B was added to tissues in a volume of 600 μl. Then, samples were loaded on Tissue Lyser (Boston Industries, Walpole, MA) and subjected to vibration and dispersal at 25 cps for 30 seconds. After 30-second cooling intervals, the 30-second homogenizations were repeated 10 times to minimize any heat generation. Samples were centrifuged at 14,000g for 15 minutes, and supernatants were collected for LC/MS/MS analysis.
Solution samples, plasma, urine, and bile were deproteinated by 1:3 dilutions with acetonitrile containing 2 μM RS138B as an internal standard. Urine samples came directly from the urinary bladder, and biliary samples were from the gall bladder. Samples were vortexed and then centrifuged at 14,000g at room temperature for 15 minutes. Supernatants were collected for LC/MS/MS analysis.
Liquid Chromatography/Mass Spectrometry Analyses
Sample aliquots, usually in 10-μl volumes, were analyzed by LC/MS/MS (Sciex API 4000). Oxime separation was performed on high performance liquid chromatography with a methanol/water gradient in 0.1% formic acid (by volume), eluted over an ACE C18-Ar column (2.1 × 100 mm; 3 μM; Mac-Mod analytical), prior to tandem mass spectrometry analysis. The transitions for RS194B and RS138B used for quantification were mass-to-charge ratio (m/z) 214 > 115 and m/z 266 > 115, respectively. The limit of detection for RS194B was ∼20 pg from the column separation.
Statistics
Statistical significance was evaluated using one-way ANOVA. P values < 0.05 were considered significant. *, **, and *** represent P values < 0.05, < 0.01, and < 0.001, respectively.
Results
Pharmacokinetics and Disposition of RS194B in Blood and Organs
Pharmacokinetics of oxime RS194B (70 mg/kg) in mouse blood, administered intramuscularly, IV (Intravenous, Retro-orbital space) RO, and intravenously (tail vein) to approximately five to eight mice, is shown in Fig. 1. When assayed at 5 and 10 minutes after antidote injection, intramuscular injection shows a slightly slower absorption and distribution of the antidote than after tail vein and retro-orbital administration. The kinetics indicate that, within 15 minutes, RS194B distributes (disperses) from the sites of injection in muscle to blood. However, after 15 minutes, the three routes of administration exhibit very similar clearance rates and elimination patterns. The equivalence of elimination kinetics from the plasma after intravenous and RO administration indicates that intravenous administrations of RS194B have low or negligible first-pass clearance by the liver. Table 1 shows the pharmacokinetic parameters of RS194B in mice blood. The apparent half-life (t1/2) in all parenteral routes is around 20 minutes. Clearance in mice appears over twice as rapid as that found in macaques (Rosenberg et al., 2017, 2018), an expected finding given the differences in cardiovascular parameters in the two animal species of diverse weights.

Pharmacokinetics of oxime RS194B (70 mg/kg) administered intramuscularly, (circle), intravenously (retro-orbital) (square), and intravenously (tail vein) (triangle). Inset is the logarithmic figure of the three administered routes (n = ∼5–8).
Plasma pharmacokinetic parameters of RS194B in mouse blood
We employed the same mouse strains to analyze antidote concentrations of RS194B in various organs and tissues (Fig. 2). We found that RS194B concentrations reach a maximum 10–15 minutes after intramuscular injection. Kidney had the highest concentration among all organs, followed by the liver. We also found extremely high concentrations of antidote in the urine. Although difficult to quantitate from urinary or gall bladder samples because of different residual bladder volumes, the elimination of RS194B appears primarily from the kidney. Urine concentrations are consistent with the high kidney tissue levels compared with other organs.

RS194B (70 mg/kg) concentrations in various tissues and tissue excretion products at designated times after intramuscular administration (blue solid circle, brain; orange solid triangle, lung; gray solid diamond, liver; yellow hollow circle, kidney; red hollow square, diaphragm; green hollow triangle, bile; purple solid square, urine) (n = 3).
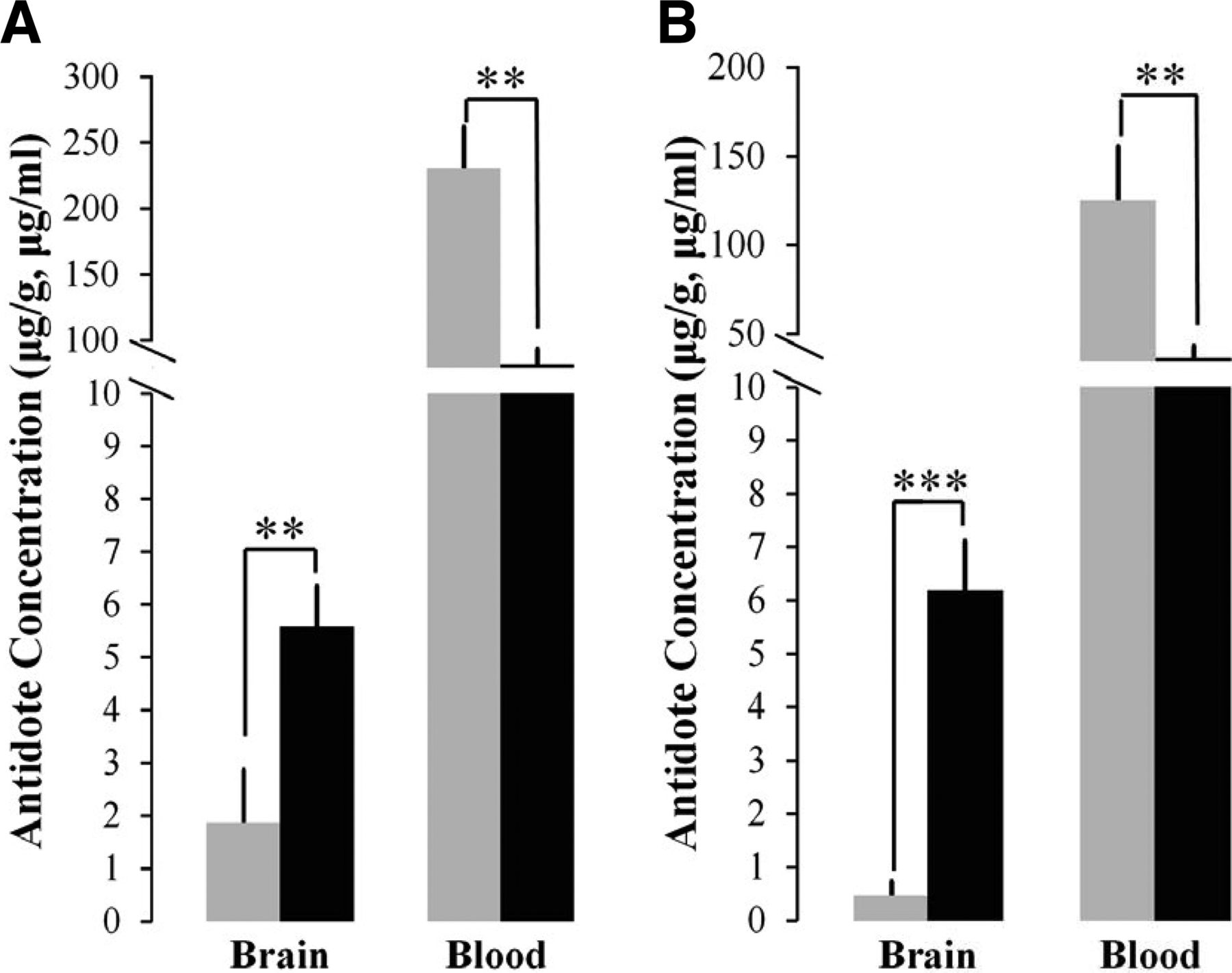
In Vivo RS194B and Pralidoxime Concentrations in Brain and Blood
In pilot experiments, RS194B and pralidoxime (2-PAM) were injected intramuscularly at the same concentration (70 mg/kg). Brain and blood samples were collected at 15 and 30 minutes after drug injection to compare the concentrations of both antidotes. Figure 3 shows that RS194B has a higher antidote concentration in brain than 2-PAM at 15 and 30 minutes after injection. These results demonstrate that RS194B has a far greater BBB penetration ratio. 2-PAM, as a quaternary amine, may be dispersed from the injection site slightly more rapidly, resulting in a higher initial blood concentration when compared with RS194B. Nevertheless, crossing of the BBB is still far greater with RS194B.

Antidote concentrations of RS194B (black) and 2-PAM (gray) in brain and blood after intramuscular injection into separated groups of mice at 15 minutes (A) and 30 minutes (B) after oxime antidote candidate injection both with 70 mg/kg (n = 3) (** P < 0.01; *** P < 0.001).
The P-gp Inhibitor TQD Increases RS194B Concentrations
Antidote concentrations in most organs and plasma increased 3- to 4-fold after prior TQD administration, whereas RS194B concentrations in gall bladder (biliary) and urine (bladder) appear to decrease when compared with the littermate controls (Fig. 4). The increases in RS194B concentrations after TQD seen in most organs in vivo presumably arise from a combination of enhanced plasma concentrations reaching and perfusing the tissue and inhibition of efflux from tissues, such as brain, kidney, and liver. Concomitantly, excretion through urine and bile appears to decrease because of TQD inhibition of the transport from the respective organs.
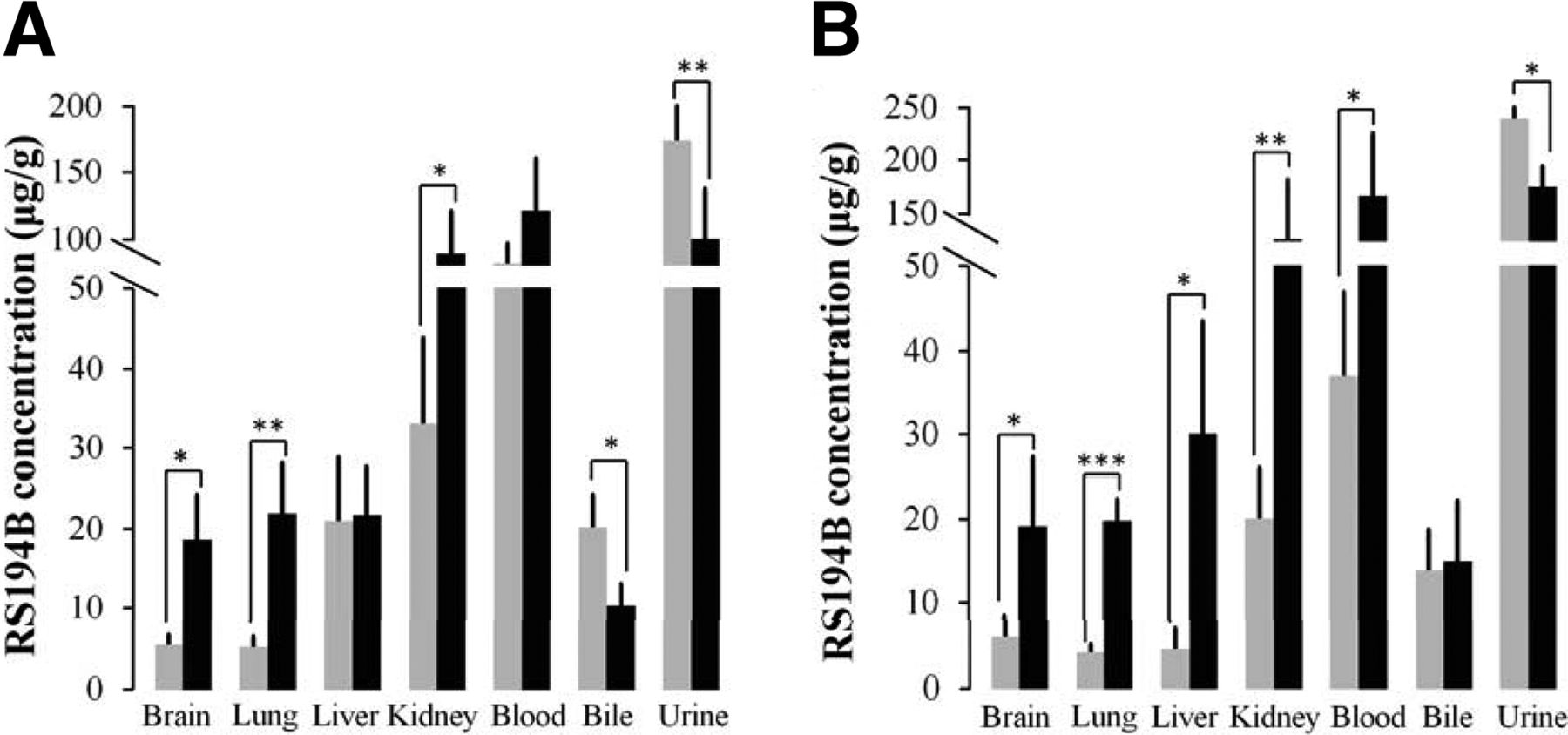
The antidote concentration of RS194B in various organs with (black) and without (gray) P-gp inhibitor TQD administration. Mice were treated with TQD (i.p., 7.5 mg/kg) 10 minutes prior to an intramuscular administration of 70 mg/kg of RS194B, and then the respective organs, blood samples, and tissue excretory fluids were collected (A) 15 minutes and (B) 30 minutes after RS194B administration (n = 3) (* P < 0.05; ** P < 0.01; *** P < 0.001).
To examine RS194B kinetics in more detail, concentrations in plasma, tissues, and excretion fluids were measured over longer time intervals. Plasma and tissue concentrations increase over the entire interval, reflecting a possible limitation from saturation on excretion of the RS194B in its various ionization states after TQD administration (Fig. 5).

Pharmacokinetics of oxime RS194B (70 mg/kg) administered intramuscularly (blue solid circle, brainbrain; pink solid triangle, lung; green solid diamond, liver; gray hollow cicle, kidney; red hollow diamond, blood; orange hollow triangle, gall bladder; purple solid square, urine) 10 minutes after P-gp inhibitor TQD injection (i.p., 7.5 mg/kg) (n = 3).
Table 2 lists the estimated pharmacokinetic parameters of RS194B in mouse blood via intramuscular injection with and without the administration of TQD. The results show that the AUC values have increased, whereas as expected, clearance values of the antidote are significantly decreased.
The pharmacokinetic parameters of RS194B in control strain mouse blood with and without prior TQD administration
The AUC has increased and Cl has decreased after the use of TQD.
Pharmacokinetic Results of RS194B in P-gp Transporter Knockout Mice
To establish the mechanism and sites of transport further, we have employed the transgenic knockout strain of mice not expressing either of the two P-gp’s in mice (Mdr1a/b). Over a period of two hours, RS194B plasma and tissue concentrations are enhanced (Fig. 6). The increases in knockout (KO) mice appear even greater than TQD administration in control mice when assessed over a 2-hour period. This may result from partial transport inhibition or slow absorption of intraperitoneal TQD. Nevertheless, our findings indicate that P-gp is the major transporter responsible for elimination of RS194B from both brain and kidney in the mouse.

Pharmacokinetics of oxime RS194B (70 mg/kg) administered intramuscularly (blue solid circle, brainbrain;, pink solid triangle, lung; green solid diamond, liver; gray hollow cicle, kidney; red hollow diamond, blood; orange hollow triangle, diaphragm gall bladder; purple solid square, urine) in (A) FVB control mice and (B) P-gp knockout mice. RS194B concentrations in P-gp knockout mice (black) and FVB control mice (gray) with statistical results are detailed for (C) 15 minutes and (D) 30 minutes after RS194B administration (n = 3) (* P < 0.05; ** P < 0.01; *** P < 0.001).
With four remaining transgenic-knockout mice, the two mice with prior TDQ administration exhibited further enhancements of RS194B brain and blood concentrations at 30 and 60 minutes (Table 3). Although the pharmacokinetics should be examined further in various animal species, transporters other than the P-gp’s will likely influence overall pharmacokinetics. It is also possible that the knockout strains express compensatory amounts of other transporters.
Combination of P-gp knockout and TQD
The drug concentration of RS194Bin P-gp KO mice with and without P-gp inhibitor TQD administration. Two of the four P-gp KO mice were treated intraperitoneally with TQD 10 min prior to 70 mg/kg, i.m., of RS194B. Then, the respective organ and blood samples were collected 30 and 60 min after RS194B injection.
Discussion
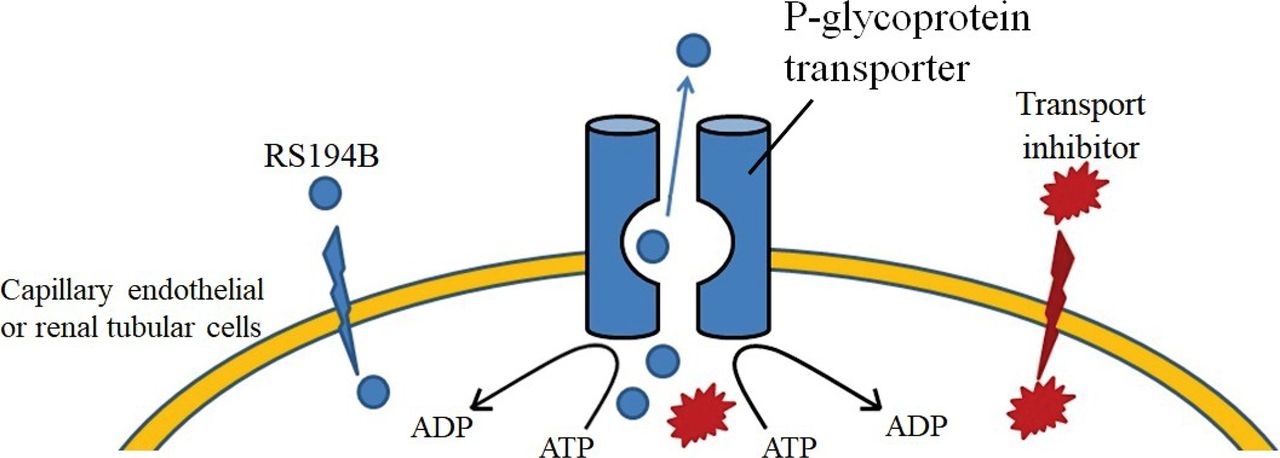
The purpose of this initial study of transporter function on RS194B elimination was to serve as a guide for BBB-permeable antidote alternatives to 2-PAM and related quaternary pyridinium aldoximes (asoxime chloride, MMB4, obidoxime) expected to be largely resistant to rapid BBB crossing (Jokanovic, 2015). For the zwitterionic oxime, with its three charged and one uncharged species (Scheme 1), both the tissue disposition and the reactivation at the OP-conjugated sites are governed by the four rapidly adjusting ionization equilibria. Both cationic pyridinium and zwitterionic oximes should be subject to efflux transport from the brain capillary endothelial cell (Scheme 2) and rapid elimination from the body (Kassa, 2002). Localized CNS capillary endothelium, extrusion transporter molecules, may critically control drug concentrations in the CNS target tissues themselves. We establish the probable dominance of the P-gp with two separate stratagems: inhibition by small molecules, using a reference P-gp inhibitor (Scheme 2) (Fox and Bates, 2007; Bankstahl et al., 2008), and deletion of the very P-gp genes expressing the two proteins (Chen et al., 2007; Geyer et al., 2009). TQD has shown success in atropine and pyridinium oxime postexposure treatment of OP toxicity in mice (Joosen et al., 2016; Meerhoff et al., 2018).

Functions of the P-gp transporter, the oxime reactivating antidote RS194B (shown in blue), and the transport inhibitor TQD (shown in red), respectively. RS194B enters the brain from the capillary by passive diffusion of the neutral species across the capillary endothelial membrane, where it is counterbalanced through extrusion by capillary endothelial transporters in the CNS, thereby serving to lower brain concentrations. Therefore, administering P-gp inhibitors such as TQD to compete with RS194B or knocking out the genes that encode the P-gp decreases RS194B extrusion and enhances its CNS (brain) concentrations.
More importantly, RS194B, as an ionizable zwitterion, confers protection to both sarin vapor and paraoxon aerosol exposures in the macaque, a nonhuman primate with a respiratory and pulmonary system resembling that of humans (Rosenberg et al., 2017, 2018). In these postexposure experiments, relatively low-dose atropine (0.28 mg/kg) was employed to reflect what might be a reasonable postexposure dose in mass terrorism settings. Head-only exposures simulate mass terrorism by a vapor or aerosol rather than parenteral (subcutaneous or intramuscular) administrations of an OP. The latter are oriented toward individualized rather than mass exposures. Accordingly, parenteral administration of OPs in rodents does not reflect a suitable mass terrorism model for rank ordering antidotes. Moreover, with OPs after inhalation acting both centrally and peripherally, the low venous return resulting from hypotension associated with OPs further limits OP parenteral administration as a model for mass terrorism (Parameswaran et al., 1999; Ockrim et al., 2006). In the prior referenced macaque studies, a relatively low dose of atropine was employed to enable the antidotal oxime to be a dominant agent in nonhuman primate exposures.
Doses of RS194B employed in mice and macaques are high (60–80 mg/kg) but scarcely out of the therapeutic realm for short-term or single-dose antidote administration. Nevertheless, a reduction of RS194B antidote doses in terms of amount and frequency of administration would be a desired strategic outcome. A P-gp inhibitor is a prime candidate for dose reduction, and we note that Joosen and colleagues have employed TQD as an exploratory agent in their cell culture studies (Joosen et al., 2016; Meerhoff et al., 2018). However, in the future, we also consider it prudent to employ other agents with P-gp inhibition capacity already approved by the FDA as a primary adjunct to the antidote (Srivalli and Lakshmi, 2012).
Accordingly, what we suggest is replacement of 2-PAM or other quaternary pyridinium aldoxime (MMB4, asoxime chloride, obidoxime, etc.) with a zwitterionic ionizable oxime, perhaps along with a transport inhibitor (Scheme 2). Hence, such an addition would simply entail repurposing an already FDA-approved agent with limited adverse pharmacologic activity. In this study, by identifying a primary target affecting drug disposition with a transport inhibitor, and then for confirmation by knocking out its encoding gene, we demonstrate the feasibility of our approach in mice.
Others have also synthesized RS194B and studied its pharmacokinetics using a radioactive tracer in the guinea pig (Malfatti et al., 2017). Their data reveal biphasic kinetics and high renal levels of RS194B. Their analysis of guinea pig pharmacokinetics disregards the initial phase in the pharmacokinetics, treating it as redistribution of the drug instead of renal elimination. As such, their volumes of distribution are much larger and their clearances are much smaller than in our analyses. We have only studied the pharmacokinetics in mice and macaques for the dual purposes of using transgenic mouse lines to understand mechanism and then exploring a primate pulmonary system resembling humans for vapor exposures.
That said, we hasten to acknowledge that mouse and macaque approaches have their respective limitations. First, TQD should be considered as a reference agent affecting antidote disposition. Second, although the P-gp knockout animal establishes an involved target transporter and the demonstration that renal and brain efflux are primary means of tissue disposition and elimination of RS194B, it is likely that other transporters are secondarily involved. Moreover, we cannot exclude the possibility that the P-gp knockout mice induce the activity of other ATP binding cassette transporter or SRC transporter cation (organic cation transporter) or organic anion transporter classification (Dresser et al., 2001; Kusuhara and Sugiyama, 2004; Roth et al., 2012). These considerations notwithstanding, it may be preferable to repurpose an FDA-approved pharmaceutical in view of a global need to find an improved antidote regimen. The mice explored in the initial data described in Fig. 6 and Table 2 with the knockout strain and TQD did not show gross signs of toxicity with respect to our antidote treatment.
Finally, we should not ignore previous studies that led to a more advanced stage of antidote development. It would have been impossible to consider all of the ionization equilibria within the active center gorge without delineating the primary (Schumacher et al., 1986) and then tertiary structures of AChE (Sussman et al., 1993), highlighting the active center, the peripheral anionic site, and the gorge leading to the active center.
An equally important consideration stems from understanding the central actions of the OPs in the compromise of cardiovascular and respiratory functions (Brezenoff and Giuliano, 1982; Buccafusco, 1996). It is critical to acknowledge experiments that highlight the CNS and, in particular, the ventral (rostral) medulla of the brain stem. Such studies point to not only a central action of OPs in AChE inhibition but also the site of action of centrally applied oxime antidotes to this regional area circumventing the BBB (Edery et al., 1986). Accordingly, an intramuscular loading dose of RS194B (coordinated with atropine in a separate injector) presents a practical acute-dosing paradigm for antidote during or after a terrorist attack initiated by an explosive device or within a controlled ventilation system.
Conclusions
We show here that concomitant use of a small-molecule P-gp inhibitor, TQD, enhances and prolongs the blood and target tissue concentrations of the ionizing zwitterionic reactivator RS194B. Establishing a mechanism for these pharmacokinetic differences is buttressed by contemporary genetics, in which a knockout strain of mice lacking the two rodent P-gp’s shows enhanced brain and blood levels of RS194B. Thus, both brain extrusion and renal elimination are inhibited and slowed. By concomitantly employing a transport inhibitor and a zwitterionic antidote, blood and brain concentrations of RS194B are enhanced, serving to augment antidote efficacy and reduce dosages and frequency of dosing.
Authorship Contributions
Participated in research design: Shyong, Samskey, Momper, Taylor.
Conducted experiments: Shyong, Sepulveda, Garcia, Samskey.
Contributed new reagents or analytic tools: Shyong, Sepulveda, Momper.
Performed data analysis: Shyong, Samskey, Radic, Sit, Sharpless, Momper, Taylor.
Wrote or contributed to the writing of the manuscript: Shyong, Taylor.
Footnotes
- Received May 5, 2021.
- Accepted June 2, 2021.
↵1 Current affiliation: School of Pharmacy and Institute of Clinical Pharmacy and Pharmaceutical Sciences, National Cheng Kung University, Tainan City, Taiwan, ROC.
This study is supported in part by the National Institutes of Health CounterACT program, NINDS, DTRA HDRA 19-1-006, and UCSD Foundation Funds. This study is also supported by Postdoctoral Research Abroad Program, Ministry of Science & Technology, Republic of China (Taiwan).
No conflict of interest exits in the submission of this manuscript, and it is approved by all authors for publication.
Abbreviations
- AChE
- acetylcholinesterase
- BBB
- blood-brain barrier
- CNS
- central nervous system
- FDA
- food and drug administration
- FVB
- Friend Virus B
- KO
- knockout
- LC/MS/MS
- liquid chromatography/mass spectrometric
- OP
- organophosphate
- 2-PAM
- pralidoxime
- P-gp
- P-glycoprotein
- t1/2
- half-life
- TQD
- tariquidar
- Vd
- volume of distribution
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}