


Abstract
The cannabinoid signaling system includes two G protein–coupled receptors, CB1 and CB2. These receptors are widely distributed throughout the body and have each been implicated in many physiologically important processes. Although the cannabinoid signaling system has therapeutic potential, the development of receptor-selective ligands remains a persistent hurdle. Because CB1 and CB2 are involved in diverse processes, it would be advantageous to develop ligands that differentially engage CB1 and CB2. We now report that GW405833 [1-(2,3-dichlorobenzoyl)-5-methoxy-2-methyl-3-[2-(4-morpholinyl)ethyl]-1H-indole] and AM1710 [1-hydroxy-9-methoxy-3-(2-methyloctan-2-yl)benzo[c]chromen-6-one], described as selective CB2 agonists, can antagonize CB1 receptor signaling. In autaptic hippocampal neurons, GW405833 and AM1710 both interfered with CB1-mediated depolarization-induced suppression of excitation, with GW405833 being more potent. In addition, in CB1-expressing human embryonic kidney 293 cells, GW405833 noncompetitively antagonized adenylyl cyclase activity, extracellular signal–regulated kinase 1/2 phosphorylation, phosphatidylinositol 4,5-bisphosphate signaling, and CB1 internalization by CP55940 (2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol). In contrast, AM1710 behaved as a low-potency competitive antagonist/inverse agonist in these signaling pathways. GW405833 interactions with CB1/arrestin signaling were complex: GW405833 differentially modulated arrestin recruitment in a time-dependent fashion, with an initial modest potentiation at 20 minutes followed by antagonism starting at 1 hour. AM1710 acted as a low-efficacy agonist in arrestin signaling at the CB1 receptor, with no evident time dependence. In summary, we determined that GW405833 and AM1710 are not only CB2 agonists but also CB1 antagonists, with distinctive and complex signaling properties. Thus, experiments using these compounds must take into account their potential activity at CB1 receptors.
Introduction
Cannabinoid receptors are part of an endogenous signaling system that is found throughout much of the body (Herkenham et al., 1990). The two canonical cannabinoid receptors, CB1 and CB2, were identified in the early 1990s (Matsuda et al., 1990; Munro et al., 1993). Cannabinoids have since been implicated in several major physiologic processes (Corcoran et al., 2015; Di Marzo et al., 2015; Alexander, 2016) and cannabinoid receptors remain a promising pharmacological target. However, a persistent hurdle has been the development of ligands that are selective for CB1 or CB2. The widespread distribution of these receptors, particularly of CB1, raises the specter of significant off-target actions, particularly if a given drug can engage both receptors. For example, it has been speculated that the analgesic activity of CB2 agonists in some preclinical pain models may be due to their concurrent activation of CB1 receptors (Manley et al., 2011). We previously reported that JWH015 [1-propyl-2-methyl-3-(1-naphthoyl)indole], a compound widely used as a selective CB2 agonist, is also a potent and efficacious CB1 agonist (Murataeva et al., 2012). In the same study, we noted that the CB2 antagonist AM630 ([6-iodo-2-methyl-1-(2-morpholin-4-ylethyl)indol-3-yl]-(4-methoxyphenyl)methanone] also blocks CB1 signaling at relatively low concentrations. The identification and careful characterization of cannabinoid receptor ligands is therefore an important task facing the cannabinoid field.
When confronted with two related receptors (e.g., activated by the same endogenous ligands), there are times when it is advantageous to not merely selectively activate one receptor, but to actively block signaling of the other receptor. Compounds with this dual quality are rare and represent an important resource. To date, the only well characterized cannabinoid receptor ligand reported to have this profile is URB447 ([4-amino-1-[(4-chlorophenyl)methyl]-2-methyl-5-phenylpyrrol-3-yl]-phenylmethanone), which is a peripherally restricted CB1 antagonist and a CB2 agonist (LoVerme et al., 2009). Even if such a compound has limited efficacy or potency, it may serve as a lead compound to allow chemists to develop novel variants. To further explore dual-action cannabinoid ligands, we examined the activity of the CB2 agonists, GW405833 [1-(2,3-dichlorobenzoyl)-5-methoxy-2-methyl-3-[2-(4-morpholinyl)ethyl]-1H-indole] and AM1710 [1-hydroxy-9-methoxy-3-(2-methyloctan-2-yl)benzo[c]chromen-6-one], toward CB1 receptors in autaptic hippocampal neurons as well as in several additional signaling assays using CB1-expressing human embryonic kidney (HEK) 293 cells or Chinese hamster ovary (CHO) cells. GW405833 is a compound that was developed as a CB2 agonist several years ago and has been used as a CB2-selective agonist in nearly 20 publications (e.g., Clayton et al., 2002; LaBuda et al., 2005; Valenzano et al., 2005; Whiteside et al., 2005). In radio-ligand binding assays, GW405833 showed high binding affinity for CB2 receptors (CHOK1 cells stably expressing human CB2), with a Ki of 3.92 ± 1.58 nM (Valenzano et al., 2005). While at CB1 receptors, GW405833 was a low-affinity ligand, with a Ki of 4772 ± 1676 nM, and was approximately 1200-fold more selective for CB2 receptors (Valenzano et al., 2005). Similarly, the structurally distinct AM1710 has been used in several publications as a CB2 agonist, mostly relating to pain research (Khanolkar et al., 2007; Rahn et al., 2011, 2014; Deng et al., 2012, 2015; Wilkerson et al., 2012). AM1710 displayed high affinity for CB2 receptors (HEK cells stably expressing human CB2 receptors), with a Ki of 6.7 nM (Khanolkar et al., 2007) and an EC50 of 11 nM (Emax of 48% ± 0.3%) to inhibit cAMP accumulation (Dhopeshwarkar and Mackie, 2016). The affinity of AM1710 for rat CB1 receptors (tested in rat brain synaptosomal membranes) was lower, with a Ki of 360 nM [95% confidence interval (95% CI), 330–390] (Khanolkar et al.,2007), and was approximately 30-fold more selective for CB2 receptors. We now report that in addition to acting as CB2 agonists, GW405833 and AM1710 also serve as antagonists at CB1 receptors, albeit with distinct pharmacological properties.
Materials and Methods
Hippocampal Culture Preparation
All procedures used in this study were carried out in accordance with and conform to the Guide for the Care and Use of Laboratory Animals adopted and promulgated by the U.S. National Institutes of Health and were approved by the Animal Care Committee of Indiana University. Mouse hippocampal neurons isolated from the CA1-CA3 region were cultured on microislands as described previously (Furshpan et al., 1976; Bekkers and Stevens, 1991). Neurons were obtained from mice (C57Bl/6, unknown sex, postnatal day 0–2) and plated onto a feeder layer of hippocampal astrocytes that had been laid down previously (Levison and McCarthy, 1991). Cultures were grown in high-glucose (20 mM) Dulbecco’s modified Eagle’s medium containing 10% horse serum, without mitotic inhibitors, and were used for recordings after 8 days in culture and for no more than 3 hours after removal from the culture medium.
Electrophysiology
When a single neuron is grown on a small island of permissive substrate, it forms synapses—or “autapses”—onto itself. All experiments were performed on isolated autaptic neurons. Whole-cell voltage-clamp recordings from autaptic neurons were carried out at room temperature using an Axopatch 200A amplifier (Axon Instruments, Burlingame, CA). The extracellular solution contained 119 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.5 mM MgCl2, 30 mM glucose, and 20 mM HEPES. Continuous flow of solution through the bath chamber (approximately 2 ml/min) ensured rapid drug application and clearance. Drugs were typically prepared as stocks and then diluted into extracellular solution at their final concentration and were used on the same day.
Recording pipettes of 1.8–3 MΩ were filled with 121.5 mM K gluconate, 17.5 mM KCl, 9 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 0.2 mM EGTA, 2 mM MgATP, and 0.5 mM LiGTP. Access resistance and holding current were monitored and only cells with both stable access resistance and holding current were included for data analysis.
Conventional Stimulus Protocol.
The membrane potential was held at –70 mV and excitatory postsynaptic currents (EPSCs) were evoked every 20 seconds by triggering an unclamped action current with a 1.0-millisecond depolarizing step. The resultant evoked waveform consisted of a brief stimulus artifact and a large downward spike representing inward sodium currents, followed by the slower EPSC. The size of the recorded EPSCs was calculated by integrating the evoked current to yield a charge value (in picocoulombs). Calculating the charge value in this manner yields an indirect measure of the amount of neurotransmitter released while minimizing the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a sampling rate of 5 kHz.
DSE Stimuli.
After establishing a 10- to 20-second 0.5-Hz baseline, depolarization-induced suppression of excitation (DSE) was evoked by depolarization to 0 mV for 50 milliseconds, 100 milliseconds, 300 milliseconds, 500 milliseconds, 1 second, 3 seconds, and 10 seconds, followed in each case by resumption of a 0.5-Hz stimulus protocol for 20–80 seconds, allowing EPSCs to recover to baseline values. This approach allowed us to determine the sensitivity of the synapses to DSE induction. To allow comparison, baseline values (prior to the DSE stimulus) were normalized to 1. DSE inhibition values are presented as fractions of 1 (i.e., a 50% inhibition from the baseline response is 0.50 ± S.E.M.). The x-axis of DSE depolarization response curves are log-scale seconds of the duration of the depolarization used to elicit DSE.
Depolarization response curves were obtained to determine pharmacological properties of endogenous 2-arachidonoylglycerol signaling by depolarizing neurons for progressively longer durations (50 milliseconds, 100 milliseconds, 300 milliseconds, 500 milliseconds, 1 second, 3 seconds, and 10 seconds). The data were fitted with nonlinear regression, allowing calculation of the effective dose or duration of depolarization at which a 50% inhibition was achieved (ED50). Statistical significance in these curves was based on nonoverlapping 95% CIs.
On-Cell Western Assay for Receptor Internalization
The internalization of the receptor was measured using an on-cell Western assay (Daigle et al., 2008). Briefly, hemagglutinin (HA)-CB1-expressing HEK cells were grown to 95% confluence in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 0.5% penicillin/streptomycin (Daigle et al., 2008). Cells were washed once with 200 μl/well HEPES-buffered saline (HBS)/bovine serum albumin (BSA; 0.08 mg/ml). Drugs in HBS/BSA were applied at the indicated concentrations to cells and were incubated for the indicated amount of time at 37°C. Cells were then fixed with 4% paraformaldehyde for 20 minutes and washed four times (200 μl/well) with Tris-buffered saline (TBS). Odyssey blocking buffer (LI-COR Inc., Lincoln, NE) was applied at 100 μl/well for 1 hour at room temperature. Anti-HA antibody (mouse monoclonal, 1:500; Covance, Princeton, NJ), diluted in 50:50 Odyssey blocking buffer and phosphate-buffered saline, was then applied for 1 hour at room temperature. Afterward, the plate was washed four times with TBS (200 μl/well). Secondary antibody (anti-mouse 680 antibody, 1:800; LI-COR, Inc.) diluted in 50:50 blocking buffer and phosphate-buffered saline was then applied for 1 hour at room temperature. The plate was then washed four times with TBS (200 μl/well). The plate was imaged using an Odyssey scanner (channel, 700 nm; intensity, 5.0; LI-COR, Inc.). Receptor internalization (expressed as the percent of basal surface levels) was calculated by dividing the average integrated intensities of the drug-treated wells by the average integrated intensities of vehicle-treated wells. (Binding of HA antibody to wild-type HEK cells was < 10% of transfected cells.) All assays were performed in triplicate, unless mentioned otherwise.
Phosphorylated Extracellular Signal-Regulated Kinase 1/2 Assay
Activation of extracellular signal-regulated kinase 1/2 (ERK1/2) was measured using an in-cell Western assay (Wong, 2004; Atwood et al., 2012). HA-CB1–expressing HEK cells were seeded onto poly-D-lysine–coated 96-well plates (75,000 cells/well) and grown overnight at 37°C in 5% CO2, humidified air. The next day, the media were replaced with HBS/BSA (0.2 mg/ml) and cells were challenged with drugs/compounds for 5 minutes at 37°C in 5% CO2, humidified air. After drug incubation, plates were emptied and quickly fixed with ice-cold 4% paraformaldehyde for 20 minutes, followed by treatment with ice-cold methanol with the plate maintained at −20°C for an additional 15 minutes. Plates were then washed with TBS/0.1% Triton X-100 for 25 minutes (five 5-minute washes). The final wash solution was then replaced with Odyssey blocking buffer (150 μl) and further incubated for 90 minutes with gentle shaking at room temperature. Blocking solution was then removed and replaced with blocking solution containing anti–phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2) antibody (1:150; Cell Signaling Technology, Danvers, MA) and was gently shaken overnight at 4°C. The next day, plates were washed with TBS containing 0.05% Tween 20 for 25 minutes (five 5-minute washes). Secondary antibody—donkey anti-rabbit conjugated with IR800 dye (Rockland, Limerick, PA), prepared in blocking solution—was added and gently shaken for 1 hour at room temperature. The plates were then washed again five times with TBS/0.05% Tween 20 solution. The plates were patted dry and scanned (channel, 700 nm; intensity, 5.5) using a LI-COR Odyssey scanner. ERK1/2 activation (expressed in percentages) was calculated by dividing the average integrated intensities of the drug-treated wells by the average integrated intensities of vehicle-treated wells. No primary antibody wells were used to determine nonspecific binding of the secondary antibody. All assays were performed in triplicate, unless mentioned otherwise.
Adenylyl Cyclase Assay
Adenylyl cyclase assays were optimized using the LANCE Ultra cAMP kit (PerkinElmer, Boston, MA) per the manufacturer’s instructions. All assays were performed at room temperature using 384-well OptiPlates (PerkinElmer). Briefly, HA-CB1 HEK cells were detached from approximately 60% confluent plates/dish using versene. Cells were then resuspended gently in 1× stimulation buffer (1× Hank’s balanced salt solution, 5 mM HEPES, 0.5 mM 3-isobutyl-1-methylxanthine, and 0.1% BSA, pH 7.4, made fresh on the day of the experiment) and were further incubated for 1 hour at 37°C in 5% CO2, humidified air. Cells were then transferred to a 384-well OptiPlate (500 cells/µl, 10 µl) and stimulated with drugs/compounds (made in stimulation buffer, 5 μl, 4× concentration, 1 μM final concentration) and forskolin (made in stimulation buffer, 5 μl, 4× concentration, 1 μM final concentration) as appropriate for 5 minutes at room temperature. Cells were then lysed by addition of 10 μl Eu-cAMP tracer working solution (4×, made fresh in 1× lysis buffer supplied with the kit; under subdued light conditions) and 10 μl ULight anti-cAMP working solution (4×, made fresh in 1× lysis buffer) and further incubated for 1 hour at room temperature. Plates were then read with the time-resolved fluorescence energy transfer mode on an Enspire plate reader (PerkinElmer).
Arrestin Recruitment Assay
Arrestin recruitment assays were performed using the PathHunter CHO-K1 CNR1 assay (CHO-mouseCB1, catalog no. 93-0959C2; DiscoverX, Fremont, CA). The assay principle is based on enzyme fragment complementation technology. In this engineered cell line, a deletion mutant of β-galactosidase is fused with arrestin and a smaller fragment of the enzyme (ProLink) is fused to the C-terminal domain of the cannabinoid receptor. The activation of the cannabinoid receptor leads to arrestin recruitment and formation of an active β-galactosidase enzyme, which then acts on substrate to emit light that can be measured on a luminescence plate reader. Cells were thawed and grown and maintained in PathHunter AssayComplete media (catalog no. 92-0018GF2; DiscoverX).
All assays were performed in poly-D-lysine–coated 96-well plates. Approximately 20,000 cells/well were plated and grown overnight at 37°C in 5% CO2, humidified air. The next day, media was replaced with 90 μl HBS/BSA (BSA, 0.2 mg/ml), an additional 10 μl of HBS/BSA containing a 10X concentration of drugs/compounds (10× concentration) was added and then incubated for 90 minutes at 37°C in 5% CO2, humidified air. For time course assays, cells were pretreated with GW405833 for the time described in the text, followed by CP55940 (2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol) plus GW405833 treatment and cell lysis. Reactions were terminated by the addition of PathHunter detection reagent (DiscoverX) and the plate was further incubated for 60 minutes at room temperature. Complementation reactions were monitored by chemiluminescence using an Enspire multiplate reader.
Inositol Phosphate 1 Assay
Accumulation of myo-inositol phosphate 1 (IP1), a downstream metabolite of IP3, was measured by using a IP-One homogeneous time-resolved fluorescence (HTRF) kit (catalog no. 62, IPAPEB; Cisbio, Bedford, MA). Functional coupling of the CB1 receptor to Gq G protein leads to phospholipase Cβ activation and initiation of the inositol phosphate (IP) cascade. Accumulated IP3 is quickly dephosphorylated to IP2 and then IP1. This assay takes advantage of the fact that accumulated IP1 is protected from further degradation by the addition of lithium chloride and IP1 levels can be easily quantified using a HTRF assay. HA-CB1 HEK cells were detached from approximately 60% confluent plates/dish using versene. Cells (10 µl, 5000 cells) were resuspended in 1× stimulation buffer (containing lithium chloride, supplied with the kit) and were incubated for 1 hour at 37°C in 5% CO2, humidified air and then transferred to a 384-well OptiPlate, followed by stimulation with drugs/compounds made in dimethylsulfoxide/ethanol as appropriate, for 10 minutes. Cells were then lysed with 5 µl IP1-d2 (made fresh in lysis buffer, supplied with the kit), followed by addition of 5 µl Ab-cryptate (made fresh in lysis buffer). Plates were incubated further for 90 minutes at room temperature and then read in HTRF mode on an Enspire plate reader. All cell-based assay experiments were performed in triplicate and were repeated at least two times, unless mentioned otherwise.
Schild Analysis
Schild plots were generated for internalization assays by employing the Schild method (Schild, 1947; Arunlakshana and Schild, 1959; Wyllie and Chen, 2007). Briefly, full concentration-response curves were obtained for CP55940 in the presence and absence of various concentrations of GW405833 or AM1710 (Figs. 3D and 4C). Next, dose ratios were calculated by dividing the half maximal effect obtained by CP55940 in the presence of a particular antagonist concentration by the half maximal effect obtained with CP55940 in the absence of antagonist. Log(dose ratio-1) was then plotted against the logarithm of antagonist concentration using linear regression (GraphPad Prism 4.0 software; GraphPad Inc., La Jolla, CA) to yield the Schild slope. A slope of 1 indicates a competitive mode of inhibition of CP55940 by a particular antagonist.
Drugs
GW405833 was obtained from Tocris Bioscience (Bristol, UK). WIN55,212-2 [(11R)-2-methyl-11-[(morpholin-4-yl)methyl]-3-(naphthalene-1-carbonyl)-9-oxa-1-azatricyclo[6.3.1.04,12]dodeca-2,4(12),5,7-tetraene] was from Sigma-Aldrich (St. Louis, MO). CP55940 was obtained through the National Institute on Drug Abuse Drug Supply Program (National Institutes of Health, Bethesda, MD). AM1710 was prepared in the laboratory of Dr. Alex Makriyannis (Department of Pharmaceutical Sciences, Center for Drug Discovery, Northeastern University, Boston, MA) (Khanolkar et al., 2007).
Results
GW405833 and AM1710 Differentially Antagonize CB1 Signaling in Autaptic Hippocampal Neurons
We first tested the effects of GW405833 and AM1710 on CB1-dependent signaling in autaptic hippocampal neurons. Depolarization of excitatory autaptic hippocampal neurons elicits a form of retrograde inhibition termed “depolarization induced suppression of excitation” or DSE (Straiker and Mackie, 2005). This can be quantified by stimulating the neuron with a series of successively longer depolarizations (50 milliseconds, 100 milliseconds, 300 milliseconds, 500 milliseconds, 1 second, 3 seconds, and 10 seconds), resulting in progressively greater inhibition of neurotransmission (Straiker et al., 2011, 2012). This yields a depolarization response curve that permits the characterization of some pharmacological properties of cannabinoid signaling, including the calculation of a median effective dose (ED50), corresponding in this case to the duration of depolarization that results in 50% of the maximal inhibition.
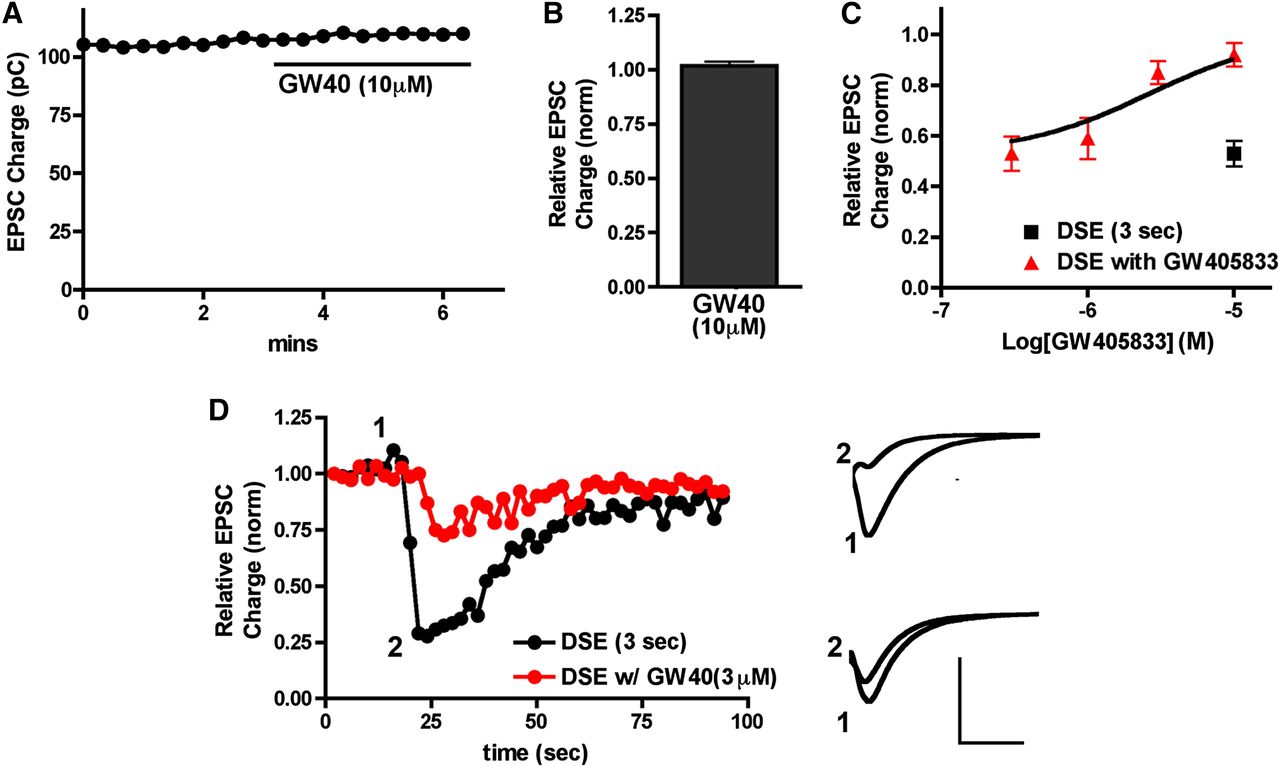
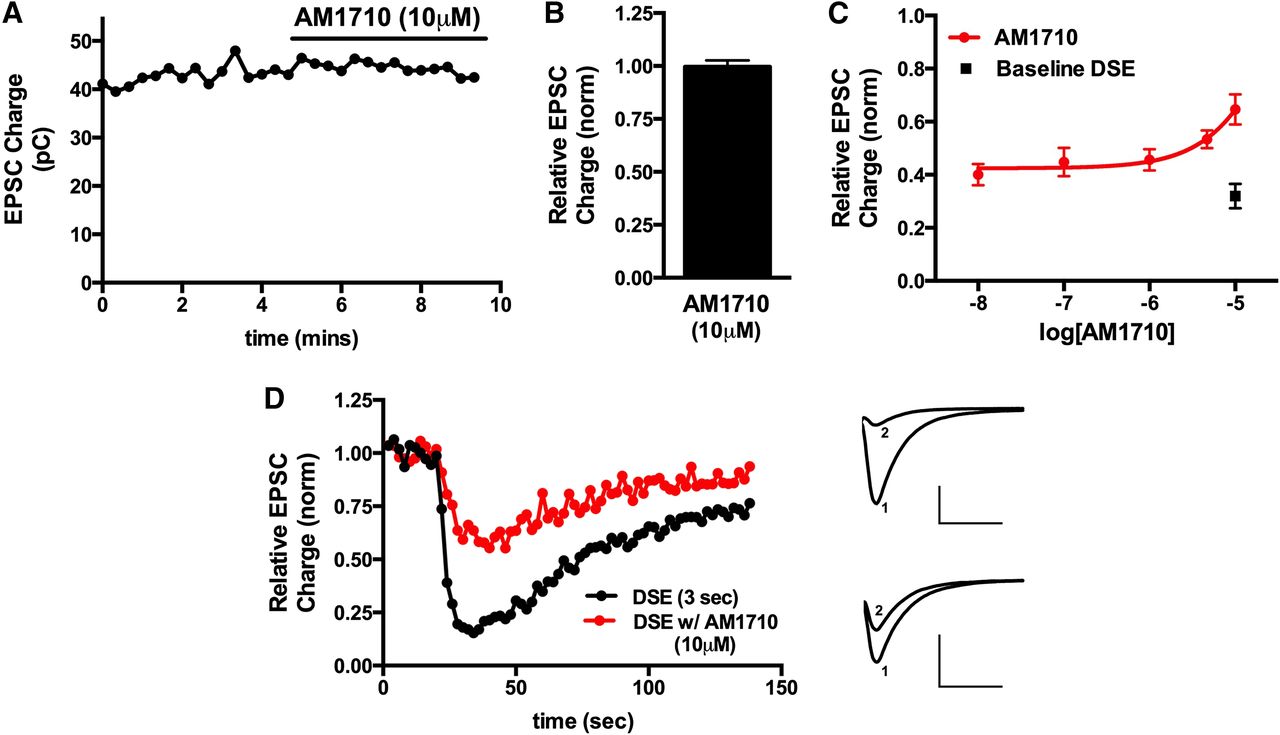
We found that although GW405833 did not directly inhibit neurotransmission (Fig. 1, A and B) (relative EPSC charge after 10 μM GW405833, 1.02 ± 0.02; n = 4), it did interfere with CB1-mediated DSE in a concentration-dependent manner, with an IC50 of 2.6 μM (Fig. 1, C and D). Similarly, AM1710 did not directly inhibit neurotransmission (Fig. 2, A and B) (relative EPSC charge after 10 μM AM1710, 1.01 ± 0.02; n = 5). However, like GW405833, AM1710 also attenuated DSE but with less efficacy and lower potency. This low potency did not allow for the calculation of an IC50 (Fig. 2, C and D).
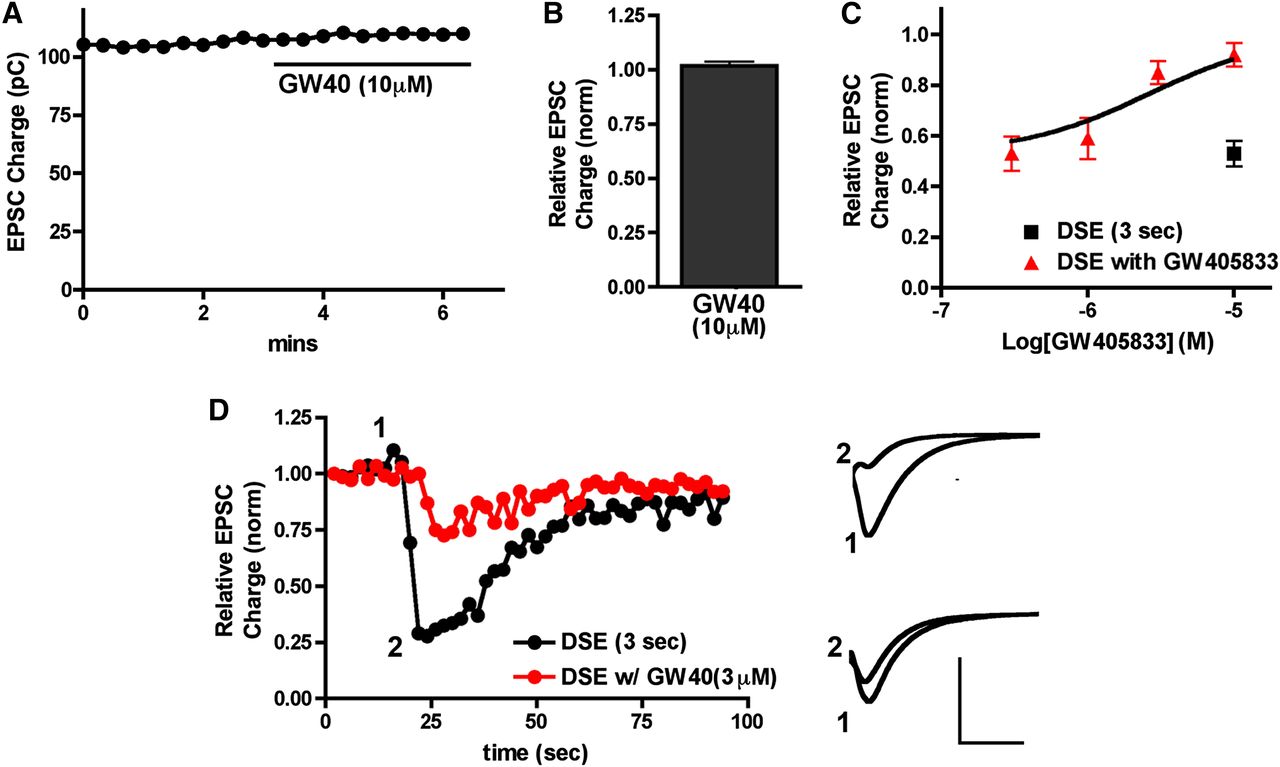
GW405833 antagonizes CB1 signaling in autaptic hippocampal neurons. (A) Sample time course shows that treatment with 10 μM GW405833 does not inhibit EPSCs. (B) Summary of data showing lack of direct inhibition of neurotransmission by GW405833 at 10 μM. (C) GW405833 inhibits CB1-dependent DSE in a concentration-dependent fashion (red triangles). Inhibition resulting from 3-second depolarization without drug is also shown (black square). (D) Sample DSE time courses before and with 3 μM GW405833 treatment. Right panels show EPSC traces at corresponding time points just before depolarization (1) and immediately after depolarization (2). Top traces are the control and bottom traces are after treatment with 3 μM GW405833. Axes: 2 nA, 30 milliseconds. GW40, GW405833; pC, picocoulomb.

AM1710 antagonizes CB1 signaling in autaptic hippocampal neurons. (A) Sample time course shows that treatment with 10 μM AM1710 does not inhibit EPSCs. (B) Summary of data showing lack of direct inhibition of neurotransmission by AM1710 at 10 μM. (C) AM1710 inhibits CB1-dependent DSE in a concentration-dependent fashion (red circles). Inhibition resulting from 3-second depolarization without drug is also shown (black square). (D) Sample DSE time courses before and with 10 μM AM1710 treatment. Right panels show EPSC traces at corresponding time points just before depolarization (1) and immediately after depolarization (2). Top traces are the control and bottom traces are after treatment with 10 μM AM1710. Axes: 2 nA, 50 milliseconds. pC, picocoulomb.
GW405833 Does Not Internalize CB1 Receptors But Antagonizes CP55940-Induced Internalization in a Concentration-Dependent Manner
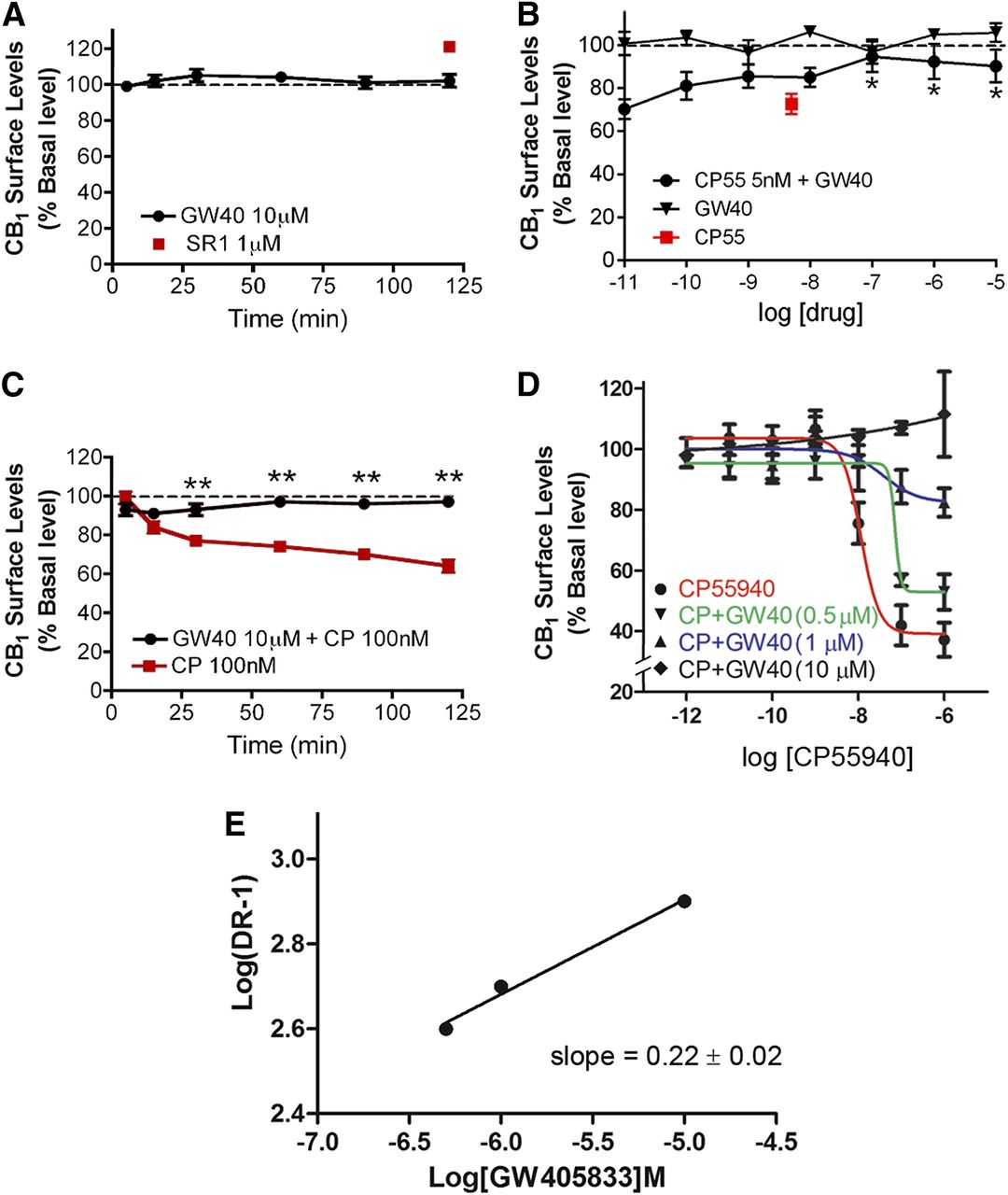
We next explored the action of GW405833 on rCB1 receptor internalization in CB1-expressing HEK293 cells in an on-cell Western assay (Daigle et al., 2008). GW405833 (10 μM) did not alter CB1 receptor surface levels over a 2-hour period (Fig. 3A), with surface levels of 102% ± 15% at 120 minutes [n = 16, P > 0.05, one-way analysis of variance (ANOVA) with Dunnett post hoc test versus baseline], suggesting that GW405833 is not an inverse agonist for CB1 receptor trafficking. The CB1 inverse agonist, SR141716 [5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide], incubated with the cells for 2 hours served as a positive control for externalization (Atwood et al., 2012), increasing CB1 surface levels by approximately 20% after 120 minutes of incubation (Fig. 3A). However, GW405833 antagonized internalization induced by 2-hour treatment with CP55940. GW405833, at concentrations ≥100 nM, antagonized internalization induced by 5 nM CP55940 (Fig. 3B), with receptor surface level values of 91% ± 7% for GW405833 (10 μM) plus CP55940 (5 nM) and 73% ± 3% for CP55940 (5 nM) (n = 24, P < 0.05, two-way ANOVA with Bonferroni post hoc test). Moreover, 10 μM GW405833 prevented internalization by a 2-hour treatment with 100 nM CP55940 (Fig. 3C), with cell surface receptor values (% of control) of 63% ± 4% for CP55940 (100 nM) and 97% ± 6.5% for CP55940 (100 nM) plus GW405833 (10 μM) at 120 minutes (n = 24, P < 0.01, two-way ANOVA with Bonferroni post hoc test). Taking together the DSE and internalization data, it appears that GW405833 is an efficacious and moderately potent antagonist at rodent CB1 receptors.

GW405833 does not internalize CB1 receptors but noncompetitively inhibits CP55940-mediated CB1 internalization. (A) Data from the on-cell Western assay show that GW405833 (10 μM) does not affect CB1 surface levels. The CB1 inverse agonist SR141716 (1 μM) reliably increases cell surface receptors and is included for comparison. (B) GW405833 diminishes CP55940-mediated CB1 internalization at higher concentrations. *P < 0.05, one-way ANOVA with Dunnett post hoc test versus CP55940 (5 nM). (C) Cotreatment with 10 μM GW405833 and 100 nM CP55940 prevents CP55940-mediated internalization. **P < 0.01, two-way ANOVA with Bonferroni post hoc for drug conditions at different time points. (D) CP55940 dose-response curves with increasing concentrations of GW405833 that were used to prepare a Schild plot (summarized in Table 1). (E) The Schild plot for GW405833 antagonism of CP55940 is consistent with noncompetitive antagonism [slope (value ± S.E.M.) ≠ 1]. Receptor internalization (expressed in % basal) was calculated by dividing the average integrated intensities of the drug-treated wells by the average integrated intensities of vehicle-treated wells (see the Materials and Methods). All assays were performed in triplicate, unless mentioned otherwise. EC50 and/or Emax values were obtained by fitting the dose-response curve using nonlinear regression with GraphPad Prism 4.0 software. A Schild plot was generated from the data plotted in Fig. 3D. Briefly, full concentration-response curves were obtained for CP55940 in the presence and absence of GW405833 at 0.5 μM, 1 μM, and 10 μM concentrations. Dose ratios (DRs) were obtained by dividing the EC50 of CP55940 obtained in the presence of various concentrations of GW405833 by the EC50 of CP55940 alone. Log(dose ratio-1) was plotted against antagonist concentrations on a logarithmic scale using linear regression (GraphPad Prism) to yield the Schild slope. CP/CP55, CP55940; GW40, GW405833; SR1, SR141716.
To explore the nature of the antagonism between GW405833 and CP55940 at CB1, we tested internalization responses for a range of GW405833 and CP55940 concentrations (Fig. 3D), sufficient to conduct a Schild analysis. As shown in Fig. 3E and Table 1, the response profile is consistent with noncompetitive antagonism.
Schild analysis for CB1 internalization is consistent with noncompetitive antagonism for GW405833 and competitive antagonism for AM1710
Data are presented with S.E.M. unless indicated otherwise.
AM1710 Does Not Internalize CB1 Receptors But Antagonizes CP55940-Induced Internalization in a Concentration-Dependent Manner
Using the same model system, we tested the effect of AM1710 in CB1 receptor internalization. AM1710 (10 μM) slightly internalized CB1 receptors after a 2-hour period (Fig. 4A) (surface levels of 93% ± 1.5% at 120 minutes; n = 16, P = 0.02, t test versus baseline), suggesting that AM1710 is a modestly efficacious agonist for CB1 receptor internalization. Furthermore, in contrast with GW405833, 10 μM AM1710 did not significantly alter the time course of internalization during a 2-hour treatment with 100 nM CP55940 (Fig. 4B), with cell surface levels (% baseline) of 62% ± 8% for CP55940 (100 nM), and 65% ± 6.7% for CP55940 (100 nM) plus AM1710 (10 μM) at 120 minutes (n = 24, P > 0.05, two-way ANOVA). In examining the effects of a range of AM1710 concentrations on CP55940-induced internalization, we found that AM1710 only modestly shifted the CP55940-response curve to the right, even at 10 μM. In addition, 20 μM and 30 μM AM1710 more substantially shifted the dose-response curve for CP55940 (Fig. 4C).

AM1710 does not internalize CB1 receptors but competitively inhibits CP55940-mediated CB1 internalization. (A) Data from the on-cell Western assay show that AM1710 (10 μM) modestly internalized CB1 receptors after 120 minutes of treatment. The CB1 inverse agonist SR141716 (1 μM) significantly increases cell surface receptors and is included for comparison. (B) In contrast with GW405833, cotreatment with 10 μM AM1710 and 100 nM CP55940 has little effect on CP55940-mediated internalization. (C) High concentrations of AM1710 decrease the potency of CP55940-mediated internalization, albeit at a higher concentration than GW405833. (D) The Schild plot for AM1710 antagonism of CP55940-induced internalization is consistent with competitive antagonism (slope ≈ 1). Receptor internalization (expressed in % basal) was calculated by dividing the average integrated intensities of the drug-treated wells by the average integrated intensities of vehicle-treated wells (see the Materials and Methods). All assays were performed in triplicate, unless mentioned otherwise. EC50 and/or Emax values were obtained by fitting the dose-response curve using nonlinear regression with GraphPad Prism 4.0 software. The Schild plot was generated from internalization assay experiments (C). Briefly, full concentration-response curves were obtained for CP55940 in the presence and absence of AM1710 at 1 μM, 3 μM, 10 μM, 20 μM, and 30 μM concentrations. Dose ratios were obtained by dividing the EC50 of CP55940 obtained in the presence of various concentrations of AM1710 by the EC50 of CP55940 alone. Log(dose ratio-1) was plotted against antagonist concentrations on a logarithmic scale using linear regression (GraphPad Prism 4.0) to yield the Schild slope. All experiments were performed in triplicate and repeated at least twice, unless mentioned otherwise. CP, CP55940; DR-1, dose ratio - 1; SR1, SR141716.
To explore the nature of the antagonism between AM1710 and CP55940 at CB1, we tested this receptor’s internalization responses for a range of AM1710 and CP55940 concentrations (Fig. 4, C and D), sufficient to conduct a Schild analysis. As shown in Fig. 4D and Table 1, the Schild analysis is consistent with a low-affinity (KB of approximately 10 μM), competitive antagonism.
GW405833 and AM1710 Attenuate Inhibition of Forskolin-Stimulated cAMP Accumulation by CP55940
We next examined whether GW405833 affected forskolin-stimulated cAMP accumulation or its inhibition by CB1 agonists in HEK cells stably transfected with rCB1. As expected, CP55940 inhibited cAMP accumulation in a concentration-dependent manner (Fig. 5A), with an EC50 of 9.5 nM and an Emax (% basal) of 45.6 ± 8.3. Although GW405833 had no effect on its own, at 1 μM it completely blocked adenylyl cyclase inhibition by CP55940 at CP55940 concentrations up to at least 1 μM (Fig. 5A). Increasing concentrations of GW405833 (300 nM, 500 nM, and 1 µM) attenuated CP55940-induced inhibition of forskolin-stimulated adenylyl cyclase (Fig. 5A). GW405833 treatment reduced the Emax (P < 0.01, t tests for 1 µM concentration) (Fig. 5A), with Emax (% basal) values of 23.2 ± 8.1 for CP55940 plus GW405833 (300 nM) and 13 ± 0.7 for CP55940 plus GW405833 (500 nM) with no significant change in the potency. EC50 values were 9.5 nM (95% CI, 2.1–19) for CP55940, 12 nM (95% CI, 3.4–18.3) for CP55940 plus GW405833 (300 nM), and 15 nM (95% CI, 4.5–23.6) for CP55940 plus GW405833 (500 nM). Classically, a reduction in Emax, with no change in potency, indicates a noncompetitive inhibition. Thus, GW405833 likely binds to a site on CB1 that is topographically distinct from that of CP55940.

GW405833 and AM1710 differentially modulate CP55940 inhibition of cAMP production and activation of pERK1/2. (A) CP55940 diminishes cAMP accumulation induced by forskolin. GW405833 has no effect on its own but increasing concentrations progressively attenuate CP55940 inhibition of cAMP accumulation and 1 μM completely blocks the action of CP55940. (B) AM1710 slightly potentiates cAMP accumulation on its own (P < 0.01, t test at 1 μM) and modestly decreases the potency of CP55940. (C) CP55940 increases pERK1/2 activation. GW405833 alone does not affect pERK1/2 levels but increasing concentrations progressively attenuate CP55940 stimulation of pERK1/2 accumulation and 1 μM completely blocks the action of CP55940. (D) For pERK1/2 activation, AM1710 alone does not affect pERK1/2 levels; however, 10 μM and 20 μM AM1710 reduce phosphorylation of ERK1/2 by CP55940 (P < 0.01, t test at 1 μM AM1710). pERK1/2 levels (expressed in %) were calculated by dividing the average integrated intensities of the drug-treated wells by the average integrated intensities of vehicle-treated wells (see the Materials and Methods). All experiments were performed in triplicate and repeated at least twice, unless mentioned otherwise. EC50 and/or Emax values were obtained by fitting the dose-response curve using nonlinear regression with GraphPad Prism 4.0 software. CP55940.
AM1710 modestly potentiated cAMP accumulation on its own (Fig. 5B), with an Emax of 117% ± 5% (P < 0.01 at 1 μM AM1710). AM1710 decreased the potency, but not the efficacy (t test at 1 µM concentration), of CP55940 inhibition of adenylyl cyclase at 10 and 20 µM (Fig. 5B), with EC50 values of 6.7 nM (95% CI, 2.3–10.1) for CP55940, 23.5 nM (95% CI, 18.8–33.3) for CP55940 plus AM1710 (10 µM), and 57.3 nM (95% CI, 45.1–77.4) for CP55940 plus AM1710 (20 µM). The decrease in potency with no effects on efficacy indicates a competitive mode of inhibition. Thus, AM1710 and CP55940 bind to the same site on CB1 receptors, leading to decreased potency of CP55940 toward CB1 receptors in the presence of AM1710.
Both GW405833 and AM1710 Attenuate CP55940 Activation of pERK1/2
Turning to pERK1/2 activation, again using HEK293 cells stably transfected with rCB1, we confirmed that CP55940 activates pERK1/2 in a concentration-dependent manner (Fig. 5C). As with cAMP experiments, GW405833 had no effect on its own, but at 1 μM it completely blocked the effects of CP55940 at CP55940 concentrations up to at least 1 μM (Fig. 5C). Increasing concentrations of GW405833 (300 nM, 500 nM, and 1 µM) inhibited CP55940-induced pERK1/2 activation, with Emax (% basal) values of 72% (95% CI, 65–73.4) for CP55940, 62% (95% CI, 58.5–63.1) for CP55940 plus GW405833 (300 nM), and 45% (95% CI, 42.4–48.2) for CP55940 plus GW405833 (500 nM). However, GW405833 did not affect the potency of CP55940 in ERK1/2 activation, with EC50 values of 9.6 nM (95% CI, 3.9–17.5) for CP55940, 13.1 nM (95% CI, 7.1–21.6) for CP55940 plus GW4059833 (300 nM), and 15.8 nM (95% CI, 8.3–27.8) for CP55940 plus GW405833 (500 nM).
AM1710 also had no effect on pERK1/2 levels on its own at 10 μM, but AM1710 at 10 μM and 20 μM progressively reduced CP55940 activation of ERK1/2 (Fig. 5D). AM1710 shifted the CP55940 concentration-response curve to the right, indicating a reduction in the potency of CP55940 for ERK1/2 activation in the presence of AM1710 (10 µM and 20 µM), with EC50 values of 4.7 nM (95% CI, 3.6–8.9) for CP55940, 34 nM (95% CI, 27.7–37.1) for CP55940 plus AM1710 (10 µM), and 47 nM (95% CI, 42.4–57.8) for CP55940 plus AM1710 (20 µM). Interestingly, increasing concentrations of AM1710 decreased the efficacy of CP55940 for ERK1/2 activation, with Emax values of 54.3 (95% CI, 50.1–59.4) for CP55950, 45.1 (95% CI, 42.2–47.6) for CP55940 plus AM1710 (10 µM), and 26 (95% CI, 19.9–32.3) for CP55940 plus AM1710 (20 µM). This mixed behavior (reduction in potency and Emax of CP55940 in the presence of AM1710) indicates mixed modes of inhibition by AM1710 in this assay.
GW405833 Time-Dependently Alters CP55940 Recruitment of Arrestin, Whereas AM1710 Does So at Relatively Lower Potency
Activation of G protein–coupled receptors often recruits β-arrestins to the cell membrane. As expected, a 90-minute treatment with CP55940 potently and efficaciously recruited arrestin in CHO-mouseCB1 cells in a concentration-dependent fashion (Fig. 6A), with an EC50 of 4.3 nM (95% CI, 2.8–6.1) and an Emax (% control) of 248 (95% CI, 233–257). Surprisingly, a 90-minute treatment with GW405833 modestly recruited arrestin on its own in a concentration-dependent fashion (Fig. 6A), with an EC50 of 0.25 nM (95% CI, 0.08–0.82; P < 0.05, one-way ANOVA with Bonferroni post hoc test) and an Emax (% control) of 46 (95% CI, 43–48). However, in contrast with other signaling pathways examined, a 5-minute pretreatment with GW405833 (1 μM) did not inhibit CP55940 recruitment of arrestin to CB1 (Fig. 6A), with an EC50 of 9.1 nM (95% CI, 3.5–16.8) and an Emax (% control) of 263 (95% CI, 249–270). We also tested a 5-minute pretreatment with 5 μM and 10 μM GW405833, finding that they too were without effect (data not shown). Interestingly, the effect of GW405833 on CP55940-mediated arrestin recruitment was time dependent. After a 20-minute pretreatment with GW405833, arrestin recruitment by CP55940 was enhanced (Fig. 6B). However, for longer GW405833 pretreatments, CP55940 recruitment was similar to recruitment without GW405833, and ultimately, starting at 60 minutes of GW405833 pretreatment, CP55940 recruitment of arrestin was inhibited by GW405833 pretreatment (Fig. 6B). The potentiation was statistically significant at 20 minutes, as were the inhibitions relative to CP55940 alone at 60 and 90 minutes (P < 0.05, one-way ANOVA with Bonferroni post hoc test). Importantly, arrestin recruitment by GW405833 alone (Fig. 6A) appeared to be time dependent, because it was not observed after 60 minutes of GW405833 treatment (Fig. 6B; P > 0.05, one-way ANOVA).
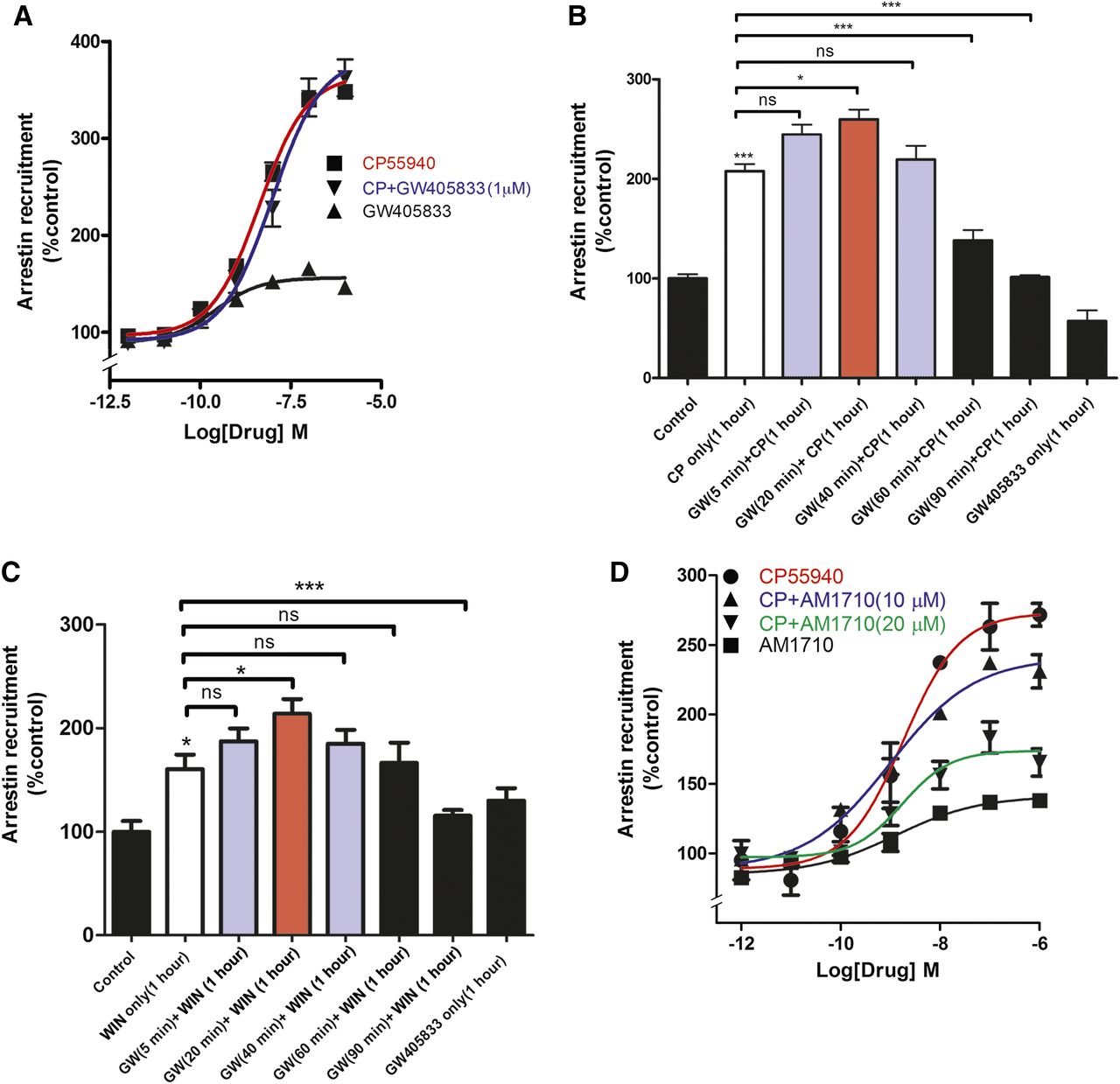
GW405833 and AM1710 differentially alter arrestin recruitment by CP55940. (A) Treatment with GW405833 (90 minutes followed by cell lysis) modestly increases arrestin recruitment in CHO-mouseCB1 cells but does not alter CP55940-induced arrestin recruitment (cells were treated with vehicle or 1 µM GW405833 for 5 minutes, followed by 1 hour of vehicle or GW405833 plus CP55940) (P < 0.01, t test at 1 μM). (B) GW405833 time-dependently alters CP55940-dependent arrestin recruitment to CB1 receptors. Brief GW405833 pretreatment (20 minutes) followed by coapplication of GW405833 with CP55940 enhances CP55940-mediated arrestin recruitment, whereas pretreatment with GW405833 for an hour or more antagonizes CP55940-mediated recruitment (P < 0.05, one-way ANOVA with Bonferroni post hoc test). (C) GW405833 alters WIN55212-2–dependent arrestin recruitment to CB1 receptors in a time-dependent fashion. Brief GW405833 pretreatment (20 minutes) followed by coapplication of GW405833 with WIN55212-2 enhances WIN55212-2–mediated arrestin recruitment, whereas pretreatment for 90 minutes antagonizes WIN55212-2–mediated arrestin recruitment (P < 0.05, one-way ANOVA with Bonferroni post hoc test). (D) AM1710 modestly increases arrestin recruitment on its own and attenuates CP55940-induced arrestin recruitment at 10 μM and 20 μM. *P < 0.05; ***P < 0.001, one-way ANOVA with Bonferroni post hoc test. All experiments were performed in triplicate and repeated at least twice, unless mentioned otherwise. EC50 and/or Emax values were obtained by fitting the dose-response curve using nonlinear regression with GraphPad Prism 4.0 software. CP, CP55940; GW, GW405833 ns, not significant; WIN, WIN55212-2.
To explore the possibility of a chemical interaction between CP55940 and GW405833 that may lead to a complex arrestin signaling profile, similar experiments were performed using the cannabinoid receptor agonist, WIN55212-2 (Fig. 6C). Again, GW405833 displayed a profile in which it modestly potentiated WIN55212-2–mediated recruitment of arrestin after 20 minutes of treatment (P < 0.05, one-way ANOVA with Bonferroni post hoc test) and then antagonized arrestin recruitment with longer treatments (90 minutes; P < 0.0001, one-way ANOVA with Bonferroni post hoc test). Thus, it appears that the biphasic stimulation/inhibition seen with GW405833 generalizes to structurally dissimilar cannabinoid receptor agonists and is not secondary to a chemical interaction between GW405833 and CP55940. One possibility is that GW405833 favors multiple/different conformations of the receptor at different time points. Another possibility is that GW405833 is a dual-steric ligand and sequentially binds to two different sites (sites distinct from the orthosteric binding site): the first site potentiates CB1 agonist-mediated arrestin recruitment, whereas the second site inhibits recruitment (e.g., Grundmann et al., 2016).
AM1710 also modestly recruited arrestin on its own (Fig. 6D), with an Emax (% control) of 138 ± 1 (P < 0.05, t test versus baseline). However, in contrast with GW405833, 5-minute pretreatment with AM1710 (10 μM and 20 μM) reduced the extent of CP55940-mediated arrestin recruitment (Fig. 6D) (P < 0.01 for 10 μM; P < 0.005 for 20 μM), without significantly affecting CP55940 potency.
GW405833 and AM1710 Attenuate WIN55212-2–Induced Increases in IP1 Levels
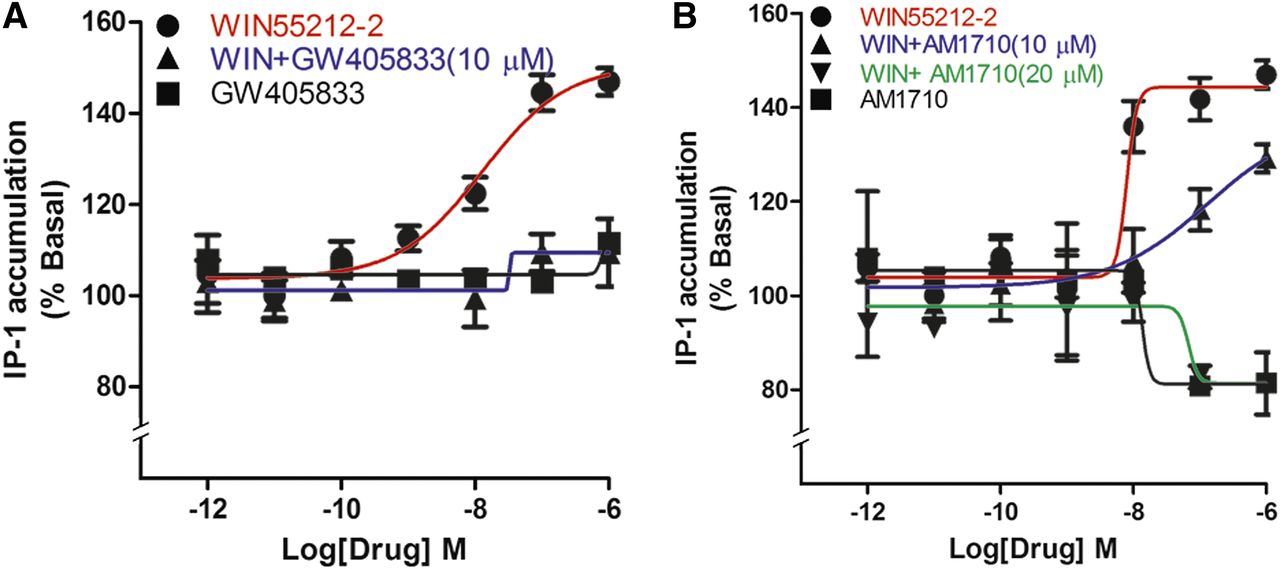
We have previously shown that certain CB1 agonists, especially aminoalkylindoles such as WIN55212-2, can engage CB1 to activate Gq signaling to increase intracellular calcium via activation of phospholipase C and release of IP3 (Lauckner et al., 2005). Therefore, we tested whether GW405833 and AM1710 affected Gq signaling in rCB1-expressing HEK cells. The CB1 agonist WIN55212-2 increased IP1 levels by approximately 50% (Fig. 7A). Pretreatment for 5 minutes with 10 μM GW405833 fully blocked the WIN55212-2 increase in IP1 (Fig. 7A), whereas GW405833 had no effect on its own. High concentrations of AM1710 alone modestly reduced IP1 accumulation (Fig. 7B), with an Emax (% basal) of 81 ± 2 (P < 0.05, t test versus baseline). As with GW405833, AM1710 attenuated the increase in IP1 elicited by WIN55212-2, doing so fully at 20 μM (Fig. 7B), with an Emax (% basal) of 81 ± 4 at 20 μM (P < 0.05, t test versus WIN55212-2).
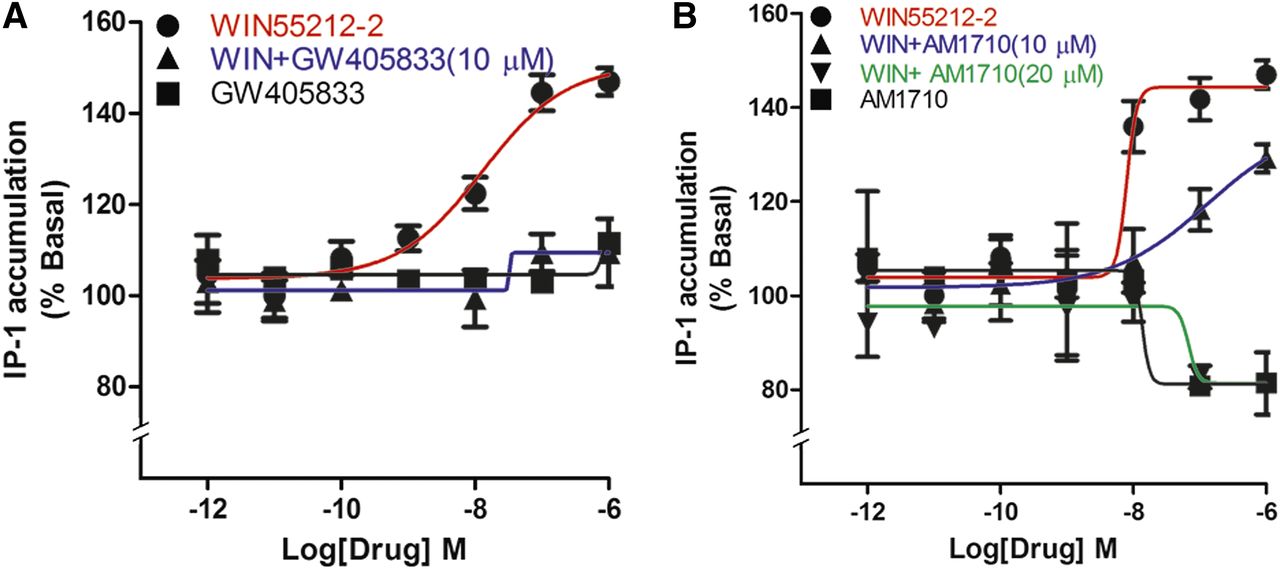
GW405833 and AM1710 block WIN55212-2 elevation of IP1 levels. (A) WIN55212-2 increased IP1 levels in a concentration-dependent manner, an effect that was fully blocked by 5-minute pretreatment with 10 μM GW405833. GW405833 had no effect on its own. (B) AM1710 had no effect on IP1 levels but did concentration-dependently block WIN55212-2–induced IP1 accumulation. IP1 levels were determined as described in the Materials and Methods (P < 0.01, t test comparing all values to IP1 accumulation after 1 μM WIN55212-2). All experiments were performed in triplicate and repeated at least twice, unless mentioned otherwise.
Discussion
Our chief finding is that GW405833 and AM1710 are not only CB2 agonists as previously reported, but they also interact with CB1 receptors with important functional consequences. These structurally distinct compounds have differential properties at CB1; most notably, our data suggest that GW405833 is a noncompetitive antagonist, whereas AM1710 is a competitive antagonist/inverse agonist at the orthosteric site for G protein signaling and a low-efficacy agonist for arrestin recruitment and internalization. AM1710 was generally less potent than GW405833, sometimes requiring 20 μM concentrations to produce a statistically significant effect. Consistent with this observation was a KB of 10 μM in the Schild analysis of internalization. The noncompetitive inhibition of CB1 signaling by GW405833 is consistent between the systems used: 1) the autaptic neurons that use the endogenous cannabinoid, 2-arachidonoylglycerol, and 2) the cell-based plate assays using transfected cells and synthetic cannabinoids. In contrast, AM1710 showed signs of pathway selectivity, with internalization and arrestin data suggesting that AM1710 is a low-potency, low-efficacy agonist for these pathways, whereas the cyclase and IP1 data are more consistent with AM1710 being a moderate-affinity inverse agonist at these pathways. Thus, the structure of AM1710 may offer an entry point for the development of arrestin-biased CB1 agonists.
These dual agonist/antagonist properties make GW405833 and AM1710 rare additions to the pharmacological toolkit available to the cannabinoid field. The only other published compound with this profile is URB447 (LoVerme et al., 2009). A compound with this profile is particularly valuable in a multidimensional system in which both CB1 and CB2 receptors are present and can potentially mediate opposing functions. For example, in the immune system where both CB1 and CB2 receptors have been found to be active, GW405833 may offer a single-drug option to dissect out the contributions of each receptor system to immune function. Another example is treatment of chronic pain, in which CB2 agonists and CB1 antagonists have both been shown to be beneficial in various preclinical models (Costa et al., 2005; Pernía-Andrade et al., 2009; Comelli et al., 2010; Gutierrez et al., 2011). It has also been suggested that the inclusion of CB1 antagonist properties in a model of neuropathic nociception would be advantageous (Rahn et al., 2008). Of course, an important question is whether the noncompetitive antagonism of CB1 receptors by GW405833 has the psychiatric liabilities associated with potent CB1 inverse agonists such as SR141716/rimonabant.
Our Schild analysis suggests that AM1710 is a low-affinity (KB of approximately 10 μM) competitive ligand at CB1. In contrast, the action of GW405833 appears to be noncompetitive in nature, suggesting that GW405833 does not bind to the orthosteric site of CB1, which is consistent with ligand binding studies (e.g., Valenzano et al., 2005). A plausible mechanism is that GW405833 acts at an allosteric site on CB1, in that case making it a negative allosteric modulator of CB1. Our results do not, however, rule out indirect action via some other receptor or signaling pathway. For example, GW405833 has also been reported to serve as a partial agonist at G protein–coupled receptor 55 and to enhance the signaling of the putative G protein–coupled receptor 55 ligand lysophosphatidylinositol (Anavi-Goffer et al., 2012), although these particular examples are unlikely in the systems studied here since GW405833 generally had little effect on its own.
GW405833 was often a potent and efficacious antagonist; in several instances, it completely blocked the effect of CP55940 at 1 μM. However, it was not as potent for the inhibition of DSE in autaptic neurons, with the relatively high IC50 of 2.6 μM. This may indicate that the interaction of GW405833 with CB1 depends on the local environment (e.g., neurons versus overexpression) or the nature and/or efficiency of receptor effector coupling in the various expression systems. GW405833 was, however, broadly efficacious, acting as an antagonist in every assay examined (albeit with a time dependence when inhibiting arrestin recruitment).
The interactions of GW405833 with CB1-mediated arrestin recruitment are quite intriguing. Brief treatment with GW405833 modestly enhanced arrestin recruitment to the CB1 receptor both in the presence and absence of CP55940. A longer treatment with GW405833 further enhanced arrestin recruitment by CP55940. However, by 1 hour, this enhancement by GW405833 shifted to a pronounced inhibition. The net inhibitory effect is consistent with the inhibitory actions seen for other signaling pathways. Transitory stimulation of arrestin signaling is also consistent with the observation that the inhibition of CP55940-mediated CB1 internalization was only evident at 30 minutes after treatment with GW405833 (Fig. 3C). This time dependence of the effects of GW405833 on arrestin recruitment was notable for several reasons. Based on our initial experiments, we would have concluded that GW405833 had no effect on arrestin recruitment by CP55940 even at 10 μM. However, those concentration-response data were collected with a 5-minute pretreatment with GW405833 followed by cotreatment with CP55940. Our results underscore the importance of considering the time course of drug actions even in relatively simple model systems (Klein Herenbrink et al., 2016). Separately, given that brief treatments were sufficient to inhibit CB1 signaling in other experiments, this raised the question of why the time dependence was limited to arrestin recruitment.
In summary, we found that the CB2 agonist GW405833 acts broadly as a medium-potency, noncompetitive CB1 antagonist. AM1710 is a low-potency, low-affinity ligand with mixed pathway-dependent, low-efficacy agonist/inverse agonist properties at CB1. Interestingly, although AM1710 appears to act competitively, GW405833 acts as a noncompetitive antagonist. The unusual pharmacological profile of either compound may prove therapeutically advantageous in certain instances. These compounds may also serve as the starting point for the development of molecules with more favorable efficacy and potency at either of the receptors while retaining duality of action.
Authorship Contributions
Participated in research design: Straiker, Mackie.
Conducted experiments: Dhopeshwarkar, Murataeva, Straiker.
Contributed new reagents or analytic tools: Makriyannis.
Performed data analysis: Dhopeshwarkar, Murataeva, Straiker.
Wrote or contributed to the writing of the manuscript: Dhopeshwarkar, Murataeva, Makriyannis, Straiker, Mackie.
Footnotes
- Received July 13, 2016.
- Accepted December 5, 2016.
This research was supported by the National Intitutes of Health Institute on Drug Abuse [Grants PO1-DA009158 (to A.M. and K.M.) and RO1-DA011322 and KO5-DA021696 and RO1-DA041229 (to K.M.)] and the National Institutes of Health National Eye Institute [Grant RO1-EY24625 (to A.S.)].
Abbreviations
- 95% CI
- 95% confidence interval
- AM1710
- 1-hydroxy-9-methoxy-3-(2-methyloctan-2-yl)benzo[c]chromen-6-one
- AM630
- [6-iodo-2-methyl-1-(2-morpholin-4-ylethyl)indol-3-yl]-(4-methoxyphenyl)methanone]
- ANOVA
- analysis of variance
- BSA
- bovine serum albumin
- CHO
- Chinese hamster ovary
- CP55940
- 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol
- DR
- dose ratio
- DSE
- depolarization-induced suppression of excitation
- EPSC
- excitatory postsynaptic current
- ERK1/2
- extracellular signal-regulated kinase 1/2
- GW405833
- 1-(2,3-dichlorobenzoyl)-5-methoxy-2-methyl-3-[2-(4-morpholinyl)ethyl]-1H-indole
- HA
- hemagglutinin
- HBS
- HEPES-buffered saline
- HEK
- human embryonic kidney
- HTRF
- homogeneous time-resolved fluorescence
- IP
- inositol phosphate
- JWH015
- 1-propyl-2-methyl-3-(1-naphthoyl)indole
- pERK1/2
- phosphorylated extracellular signal-regulated kinase 1/2
- rCB1
- ratCB1
- SR141716
- 5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide
- TBS
- Tris-buffered saline
- URB447
- [4-amino-1-[(4-chlorophenyl)methyl]-2-methyl-5-phenylpyrrol-3-yl]-phenylmethanone
- WIN55,212-2
- (11R)-2-methyl-11-[(morpholin-4-yl)methyl]-3-(naphthalene-1-carbonyl)-9-oxa-1-azatricyclo[6.3.1.04,12]dodeca-2,4(12),5,7-tetraene
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}