


Abstract
The airway epithelium is critical in the pathogenesis of chronic inflammatory diseases, such as asthma and chronic obstructive pulmonary disease, and, by expressing numerous inflammatory genes, plays a prominent role in disease exacerbations. Since inflammatory gene expression often involves the transcription factor nuclear factor (NF)-κB, this signaling pathway represents a site for anti-inflammatory intervention. As the airway epithelium is targeted by inhaled therapeutic agents, for example corticosteroids, human A549 pulmonary cells and primary human bronchial epithelial (HBE) cells were selected to evaluate inhibitor of κB kinase (IKK) inhibitors. In A549 cells, interleukin (IL)-1β and tumor necrosis factor (TNF) α increased phosphorylation of IκBα, and this was followed by loss of IκBα, induction of NF-κB DNA binding, and the induction of NF-κB-dependent transcription. These events were repressed by the IKK-selective inhibitors, PS-1145 [N-(6-chloro-9H-β-carbolin-8-ly) nicotinamide] and ML120B [N-(6-chloro-7-methoxy-9H-β-carbolin-8-yl)-2-methyl-nicotinamide]. Inhibition of NF-κB-dependent transcription was concentration-dependent and correlated with loss of intercellular adhesion molecule (ICAM)-1 expression. Similarly, IL-1β- and TNFα-induced expression of IL-6, IL-8, granulocyte macrophage-colony-stimulating factor (GM-CSF), regulated and activation normal T cell expressed and secreted (RANTES), growth-related oncogene α, and monocyte chemotactic protein-1 (MCP-1) was also significantly repressed. Likewise, PS-1145 and ML120B profoundly reduced NF-κB-dependent transcription induced by IL-1β and TNFα in primary HBE cells. Parallel effects on ICAM-1 expression and a significant repression of IL-8 release were observed. In contrast, the corticosteroid, dexamethasone, was without effect on NF-κB-dependent transcription or the expression of ICAM-1. The above data provide strong support for an anti-inflammatory effect of IKK2 inhibitors acting on the pulmonary epithelium and suggest that such compounds may prove beneficial in situations where traditional corticosteroid therapies prove inadequate.
The airway epithelium plays a key role in diseases such as asthma and chronic obstructive pulmonary disease (COPD) and their exacerbations via the production of numerous cytokines, chemokines, inflammatory enzymes, adhesion molecules, and other mediators (Schwiebert et al., 1996; Bousquet et al., 2000; Proud and Chow, 2006). Thus, classic proinflammatory cytokines, including interleukin (IL)-1β, tumor necrosis factor (TNF) α, and IL-6, stimulate the inflammatory response by enhancing cell activation and the further expression of inflammatory genes. Likewise, epithelial production of chemokines, including IL-8, regulated and activation normal T cell expressed and secreted (RANTES), monocyte chemotactic protein (MCP)-1, and growth-related oncogene (GRO) α, promotes the chemotaxis of neutrophils, eosinophils, and monocytes, which may variously contribute to the pathogenesis of both asthma and COPD. Likewise, the enhanced expression of adhesion molecules, for example intercellular adhesion molecule (ICAM)-1, on the surface of epithelial cells will also promote the recruitment of inflammatory cells to the airways and is therefore proinflammatory.
Importantly, the clinical presentation of both asthma and COPD is characterized by acute exacerbations, which are often the result of specific insult to the airways. In this regard, the epithelium plays a critical role because these cells are among the first to be targeted by airborne insults, allergens, noxious chemicals, other pollutants including diesel particulates, as well as by viruses and bacteria, which are well established as major causative agents of disease exacerbations (Bousquet et al., 2000; Proud and Chow, 2006). In addition, inhaled medications, for example, anti-inflammatory corticosteroids (glucocorticoids), may also have impact primarily on the airway epithelium, and this further highlights the importance of this cell type in respect to the therapeutic management of both asthma and COPD (Barnes, 2001). Although most asthmatics are adequately controlled by inhaled corticosteroids, there are individuals who, due to disease severity or steroid insensitivity, respond poorly to such treatments and therefore require doses that will invoke undesirable side effects (Adcock and Ito, 2004; Barnes, 2004). Likewise, inhaled corticosteroids are only marginally effective in COPD and may be of lesser benefit in viral exacerbations of asthma (Barnes and Hansel, 2004; Edwards et al., 2006). Taken together, these findings suggest an urgent need for novel anti-inflammatory therapies in the treatment of airway inflammatory diseases such as asthma and COPD (Barnes, 2004; Barnes and Hansel, 2004).
The transcription factor, nuclear factor (NF)-κB, is a major regulator of inflammatory genes, including cytokines, chemokines, inflammatory enzymes, adhesion molecules, and others (Barnes and Karin, 1997), and is therefore considered as a target for novel anti-inflammatory therapies (Karin et al., 2004). NF-κB typically consists of heterodimers of p50 (NFκB1) and p65 (RelA) and becomes activated following the signal-induced phosphorylation and degradation of inhibitory IκB proteins. Loss of IκBα releases NF-κB, which then translocates from the cytoplasm to the nucleus, where it participates in the activation of inflammatory genes. The identification of the IκB kinase (IKK), IKK2, as being the principal kinase responsible for the phosphorylation of serine residues 32 and 36 of IκBα has lead to the development of small-molecule inhibitors of this kinase (Karin et al., 2004). In the current study, we have examined pulmonary A549 cells and primary human bronchial epithelial (HBE) cells to evaluate the pharmacological utility of inhibiting this pathway using the novel IKK-selective inhibitors PS-1145 (Hideshima et al., 2002; Castro et al., 2003) and ML120B (Nagashima et al., 2006; Wen et al., 2006).
Materials and Methods
Reagents. IL-1β and TNFα were from R&D Systems (Abingdon, UK or Hornby, ON, Canada). The IKK2-selective inhibitors PS-1145 and ML120B have been described previously (Hideshima et al., 2002; Castro et al., 2003; Nagashima et al., 2006; Wen et al., 2006). Both PS-1145 and ML120B were supplied as free bases by Millennium Pharmaceuticals (Cambridge, MA) and were dissolved in dimethyl sulfoxide (DMSO) before dilution in tissue culture medium. All other reagents were from Sigma (Poole, UK) unless otherwise stated.
Cell Culture and Adenovirus Infection. Human A549 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum as described previously (Catley et al., 2005). Normal human lung tissue was obtained via a tissue retrieval service and with appropriate ethical approval. Primary HBE cells were harvested and cultured in bronchial epithelial cell growth medium (Bio Whittaker, Walkersville, MD) as described previously (Proud et al., 2004). In all cases, cells, A549 or primary HBE, were incubated overnight in basal medium, without either fetal calf serum or supplements before drug treatments or stimulation.
The null, dominant IκBαΔN, and dominant-negative IKK1(KM) and IKK2(KA) expressing adenoviruses have all been described previously (Krappmann et al., 1996; Zandi et al., 1997; Catley et al., 2003, 2005). All viruses were titrated by endpoint dilution and plaque assay to determine plaque-forming units. Viruses were diluted in Dulbecco's modified Eagle's medium (Sigma) to a multiplicity of infection (MOI) of 10 before infection of A549 cells. This dose of virus showed no effect on cell viability (Catley et al., 2005). After adenoviral vector infection, A549 cells were incubated for 24 h. The cells were then serum-starved for an additional 24 h before treatments with drugs and/or cytokines.
Analysis of NF-κB-Dependent Transcription. A549 cells harboring a stable NF-κB-dependent luciferase reporter (6κBtk.neo) that contains six copies of a consensus NF-κB binding motif were as described previously (Bergmann et al., 1998). NF-κB reporter assays in primary HBE cells were conducted using an adenovirus carrying an NF-κB-dependent luciferase reporter (Ad-NF-κB-luc). This construct contains five copies of the classic NF-κB enhancer (5′-TGG GGA CTT TCC GC-3′), a TATA box and luciferase gene from pNF-κB-luc (Stratagene, La Jolla, CA) in an Ad5 vector (Catley et al., 2006). After infection of HBE cells at a MOI of ∼5 and incubation for 24 h in media without growth supplements, the cells were treated with drugs and then stimulated with IL-1β or TNFα. Both primary HBE and A549 cells were harvested after 8 h, and luciferase activity was determined using a commercial kit (Promega, Madison, WI).
Electromobility Shift Assays and Luciferase Assay. Electrophoretic mobility shift assay (EMSA) for NF-κB was performed using a double-stranded 32P-radiolabeled probe containing the consensus NF-κB motif (underlined) (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) (Promega) and nuclear extracts as described previously (Nasuhara et al., 1999).
Western Blotting and Cytokine Release Measurements. Detection of proteins by Western blot analysis was carried out using commercial antibodies to serine 32 and 36 phosphorylated IκBα (5A5) (Cell Signaling, Danvers MA), total IκBα (sc371) (Santa Cruz, Santa Cruz, CA), ICAM-1 (Sc8439) (Santa Cruz), and GAPDH (6G5) (Biogenesis, Poole, Dorset, UK) as described previously (Nasuhara et al., 1999; Catley et al., 2005). Measurement of cytokine release was by SearchLight Proteome Array sandwich ELISAs using the Pierce custom service (Perbio, Woburn, MA). IL-6 and IL-8 measurements were also performed using a commercial ELISA kit (R&D Systems).
Statistical Analysis. All values are expressed as means ± S.E.M. Statistical significance was determined using one-way analysis of variance with a Bonferroni post test for comparison with the control sample. Significance was taken where P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Results
Effect of Dominant IκBαΔN and Dominant-Negative IKK1 and IKK2 Overexpression on the Expression of IL-6. To examine the roles of NF-κB, IKK1, and IKK2 in the induction of inflammatory gene expression by IL-1β and TNFα, A549 cells were infected with adenoviral vectors able to direct overexpression of IκBαΔN, a dominant inhibitor of NF-κB, or dominant-negative versions of IKK1 and IKK2 (Catley et al., 2005). In these studies, IκBαΔN and dominant-negative IKK2, but not dominant-negative IKK1 or an empty (null) virus, prevented the induction of NF-κB DNA binding and the induction of NF-κB-dependent transcription in A549 cells (Catley et al., 2005). This previous study documented a parallel inhibitory effect on the expression of ICAM-1, GM-CSF, and IL-8. In the current investigation, we have reexamined the supernatants from this earlier study and now extend our observations to include IL-6 (Fig. 1). Thus, IL-6 release was substantially increased by both IL-1β and to a lesser extent by TNFα. In each case, prior infection with the null adenovirus showed no significant effect, whereas dominant IκBαΔN reduced IL-6 release to near-background levels. Likewise, the dominant-negative IKK2 adenovirus prevented IL-6 release, whereas dominant-negative IKK1 was without effect suggesting that IL-6 is both NF-κB- and IKK2-dependent.

Dominant IκBαΔN and dominant-negative IKK2 prevent expression of IL-6 in A549 cells. A549 cells were either not infected or infected with the indicated adenoviruses at a MOI of 10. Cells were incubated in serum free medium for 24 h before either no stimulation or stimulation with IL-1β (1 ng/ml) or TNFα (10 ng/ml). After a further 18 h, supernatants were assayed for the presence of IL-6. Data (n = 6) are plotted as means ± S.E.M. Null represents empty Ad5 vector, and IκBαΔN, dnIKK1, and dnIKK2 represent Ad5 vector overexpression of IκBαΔN, dominant-negative IKK1, and dominant-negative IKK2, respectively.

PS-1145 and ML120B prevent IL-1β- and TNFα-induced phosphorylation of IκBα. A549 cells were preincubated, or not, with various concentrations of PS-1145 (A) or ML120B (B) or 0.1% (v/v) DMSO. After 1.5 h, cells were stimulated with either IL-1β (1 ng/ml) or TNFα (10 ng/ml). Two minutes later, cells were harvested for Western blot analysis of serine 32 and 36 phosphorylated IκBα (P-IκBα) and total IκBα (IκBα). Blots representative of two to four determinations are shown. After densitometric analysis, data were expressed as a ratio of P-IκBα/IκBα and are plotted as means ± S.E.M.
PS-1145 and ML120B Inhibit Activation of NF-κBin A549 Cells. To explore the potential utility of IKK inhibitors as novel anti-inflammatory agents acting on the pulmonary epithelium, the effects of the two small-molecule IKK inhibitors, PS-1145 and ML120B (Hideshima et al., 2002; Castro et al., 2003; Wen et al., 2006), were tested on the activation of NF-κB by both IL-1β and TNFα. Since activation of NF-κBin A549 cells is dependent on IKK2, which phosphorylates serines 32 and 36 of IκBα as a prelude to IκBα degradation and activation of NF-κB (Catley et al., 2005), the effect of both PS-1145 and ML120B was examined on these parameters. In A549 cells, IKK kinase activity is maximal within 5 min of TNFα stimulation, and both IL-1β and TNFα lead to substantial losses of IκBα within this time frame (Newton et al., 1998a; Catley et al., 2004). Therefore, the effect of PS-1145 and ML120B on signal-induced phosphorylation of IκBα was examined 2 min poststimulation, i.e., at a time where little IκBα degradation had occurred (Fig. 2). In resting cells, low or undetectable levels of serine 32 and 36 phosphorylated IκBα were detected. However, phosphorylation of serines 32 and 36 was readily detectable following either IL-1β or TNFα treatment. Prior incubation with PS-1145 or ML120B led to a concentration-dependent inhibition of phosphorylated IκBα, and this is consistent with inhibition of IKK2 (Fig. 2).
Likewise, before treatment of A549 cells with PS-1145 or ML120B markedly reduced both IL-1β- and TNFα-induced degradation of IκBα (Fig. 3A). Parallel analysis of NF-κB DNA binding by EMSA in nuclear extracts derived from these same experiments revealed that the prevention of signal-induced degradation of IκBα was also associated with reduced levels of NF-κB DNA binding induced by either IL-1β or TNFα (Fig. 3B). Given selectivity for IKK2 (Hideshima et al., 2002; Castro et al., 2003; Nagashima et al., 2006; Wen et al., 2006), the data presented in Figs. 2 and 3 are consistent with IKK2 being the kinase responsible for activation of NF-κB following the phosphorylation and loss of IκBα.

PS-1145 and ML120B attenuate stimulus-induced loss of IκBα and the induction of NF-κB DNA binding activity. A549 cells were pretreated for 1.5 h with the indicated concentrations of PS-1145 or ML120B. Cells were either not stimulated or stimulated with IL-1β (1 ng/ml) or TNFα (10 ng/ml) and harvested after 5 min. A, Western blot analysis of total IκBα and GAPDH was performed on cytoplasmic extracts. B, NF-κB DNA binding activity was analyzed by EMSA on nuclear extracts. Binding reactions were carried out in the presence of a 100-fold excess of cold probe (XS), and specific binding complexes are indicated. All blots are representative of three such experiments.
To confirm that these events equate to effects on NF-κB-dependent transcription, PS-1145 and ML120B were tested on NF-κB-dependent transcription using an NF-κB-dependent luciferase reporter that had been established previously and validated as a stable A549 cell line (Catley et al., 2005) (Fig. 4). Pretreatment with either PS-1145 or ML120B produced a concentration-dependent inhibition in the luciferase activity induced by either IL-1β (EC50, 1.4 and 8.2 μM, respectively) or TNFα (EC50, 4.3 and 6.6 μM, respectively) (Fig. 4). In each case, the EC50 values and inhibition characteristics correlate with the effects observed on serine 32 and 36 phosphorylated IκBα.
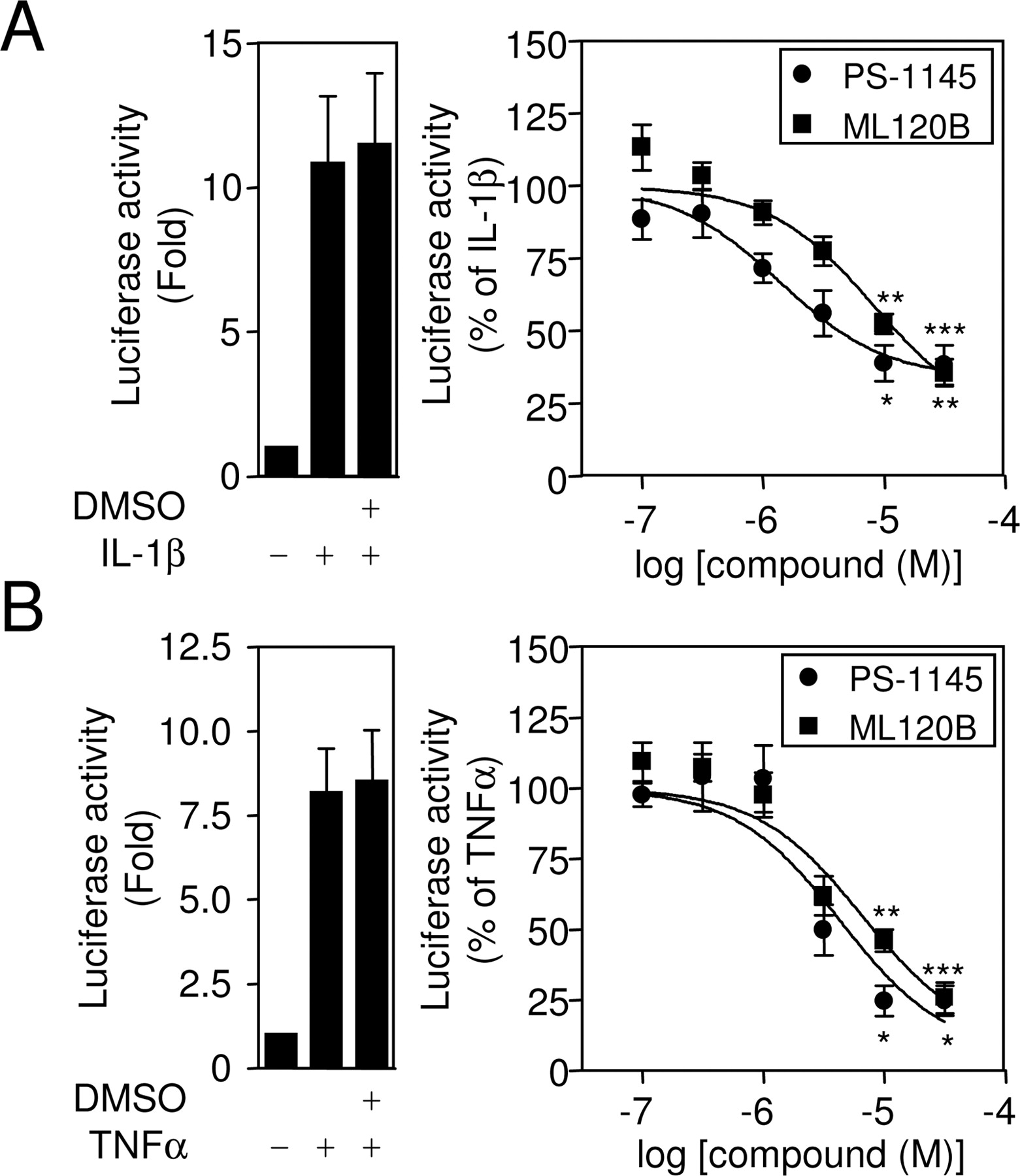
Concentration-dependent repression of NF-κB-dependent transcription by PS-1145 and ML120B. A549 cells harboring a stable NF-κB-dependent luciferase reporter (6κBtk.neo) were stimulated with IL-1β (1 ng/ml) (A) or TNFα (B) (10 ng/ml) in the presence or absence of a 1.5-h preincubation with either 0.1% (v/v) DMSO (left) or various concentrations of either PS-1145 or ML120B (right). After 8 h, cells were harvested for luciferase activity determination. In each case, the left panels show data (n = 16–17) expressed as -fold induction, whereas right panels (n = 7–9) show the effect of PS-1145 and ML120B, and the data are expressed as a percentage of stimulated. All data are plotted as means ± S.E.M.
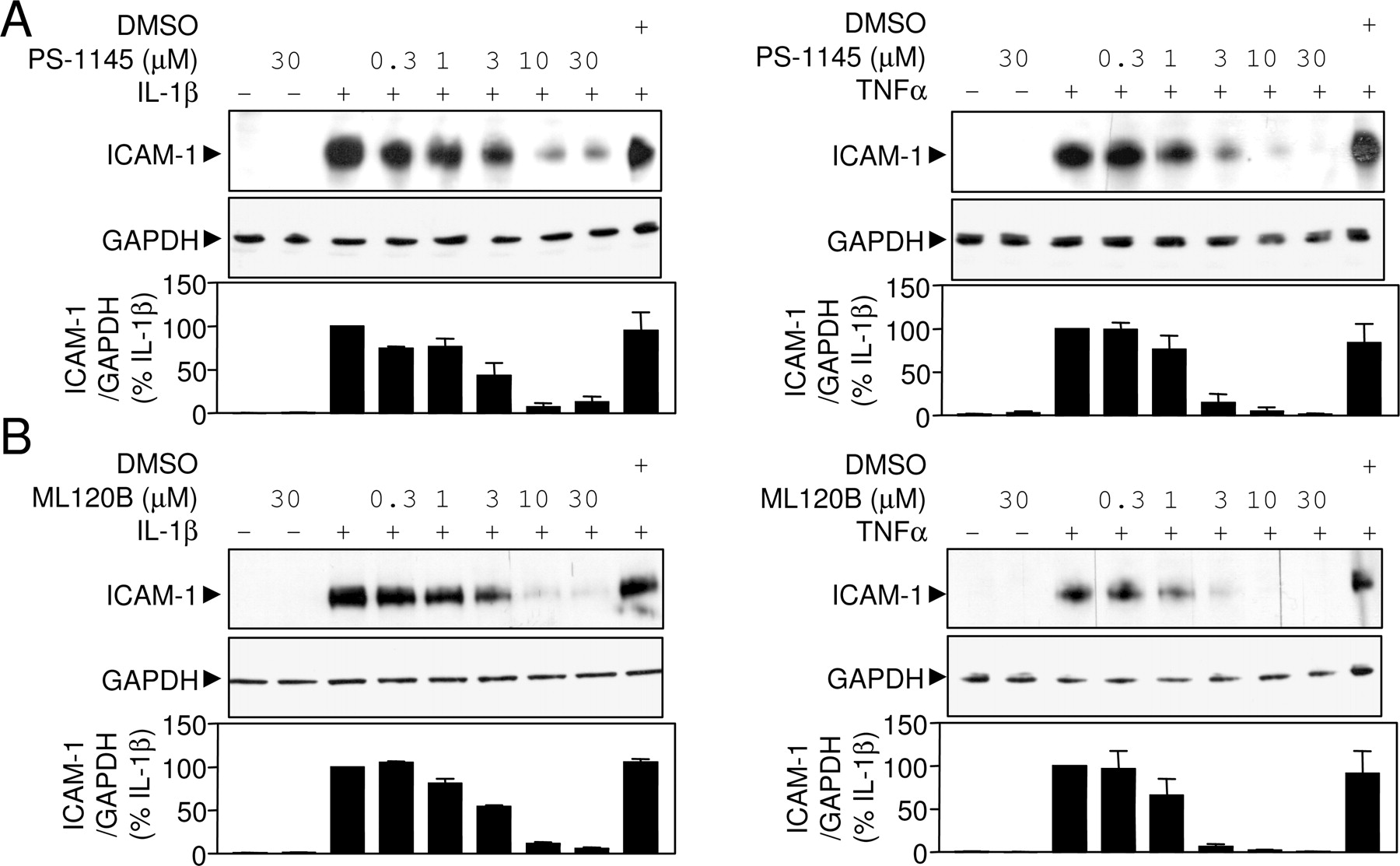
Effect of PS-1145 and ML120B on Inflammatory Gene Expression in A549 Cells. To examine the effects of PS-1145 and ML120B on proinflammatory gene expression, ICAM-1 was selected as a physiologically relevant but highly NF-κB-dependent target gene (Catley et al., 2005). Western blot analysis for ICAM-1 in the cell extracts obtained from the experiments depicted in Fig. 4 revealed a profound inducibility by both IL-1β and TNFα (Fig. 5). Both PS-1145 and ML120B produced concentration-dependent reductions in ICAM-1 expression to near-background levels (Fig. 5). This effect correlated with the inhibition of NF-κB-dependent transcription and is therefore consistent with a causal relationship.
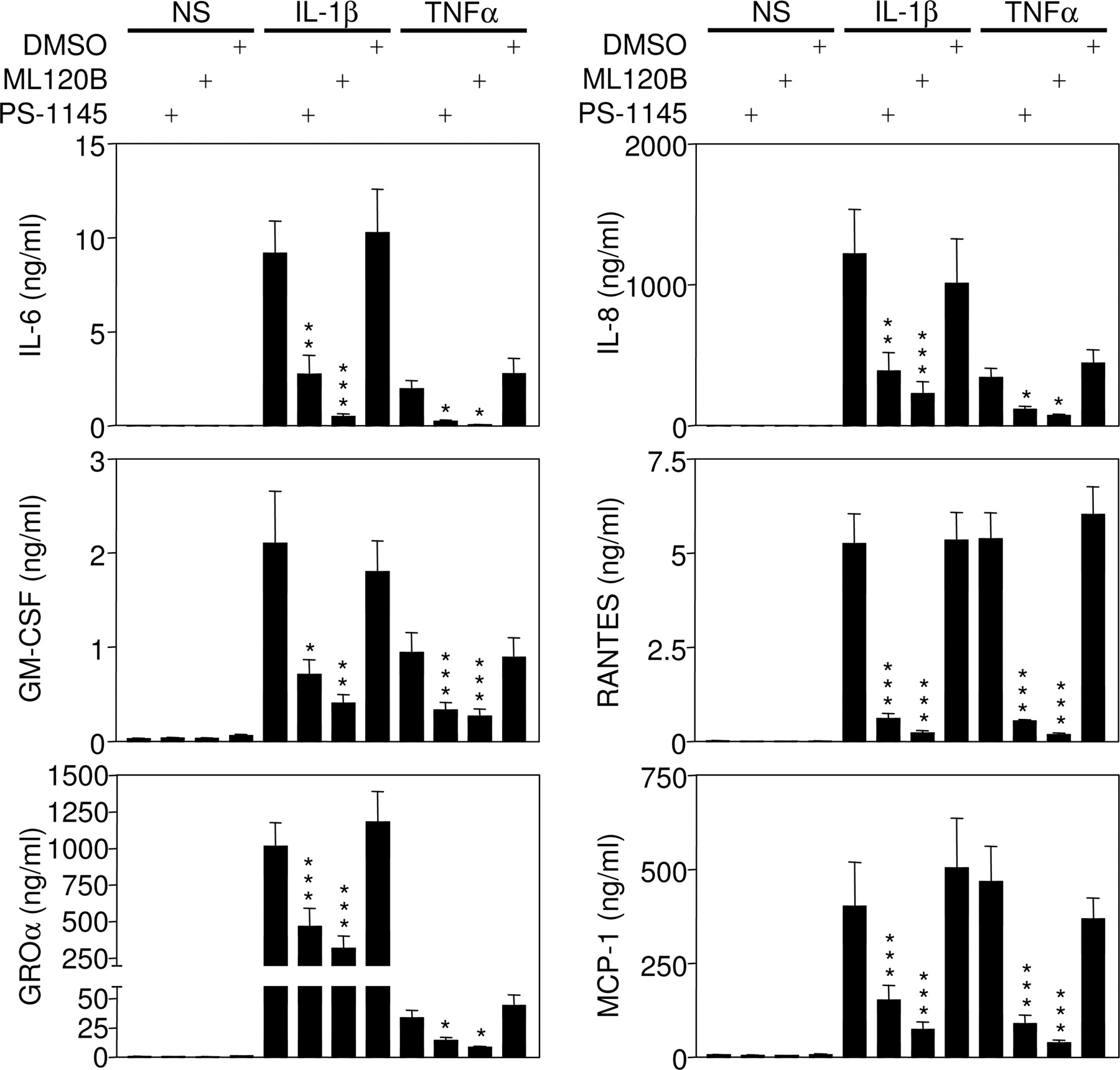
To examine the potential for small-molecule IKK inhibitors to prevent the expression of further inflammatory genes, A549 cells were treated with near-maximal effective concentrations of either PS-1145 or ML120B before stimulation with either IL-1β or TNFα. After 18 h, total cellular lysates and supernatants were harvested. Initially, Western blot analysis was performed for ICAM-1. As was described in Fig. 5, both PS-1145 and ML120B near totally prevented ICAM-1 expression induced by IL-1β and TNFα (data not shown) and therefore confirmed the efficacy of the treatments in this experiment. The supernatants from this experiment were therefore examined for expression of IL-6, IL-8, GM-CSF, RANTES, GROα, and MCP-1 (Fig. 6). Similar levels of RANTES and MCP-1 release were induced by IL-1β and TNFα treatments. However, the induction of IL-6, IL-8, and GM-CSF by TNFα was ∼50 to 80% lower than for IL-1β, whereas TNFα-induced GROα was some 35- to 40-fold lower than that for IL-1β. In all instances, preincubation with PS-1145 or ML120B resulted in a significant attenuation of cytokine release (Fig. 6). Because inhibition of NF-κB may promote apoptosis in a variety of cells, including in A549 cells (Catley et al., 2005), cell viability was also monitored (see supplemental data). Thus, 18 h of treatment with IL-1β and TNFα in the presence or absence of PS-1145 or ML120B at concentrations of up to 30 μM showed no effect on cell viability (Supplemental Table 1; Supplemental Fig. 1).

Concentration-dependent inhibition of ICAM-1 expression by PS-1145 (A) and ML120B (B). After the indicated treatments, cellular lysates from the experiments depicted in Fig. 4 were subjected to Western blot analysis of ICAM-1 and GAPDH. For each set of conditions, three or four blots were conducted, and representatives are shown. After densitometric analysis, ICAM-1 data were normalized to GAPDH, expressed as a percentage of stimulated, and plotted as means ± S.E.M.

PS-1145 and ML120B inhibit the release of proinflammatory cytokines and chemokines induced by IL-1β or TNFα. A549 cells were preincubated with PS-1145 (10 μM), ML120B (10 μM), or 0.1% (v/v) DMSO for 1.5 h before either no stimulation or stimulation with IL-1β or TNFα. After 18 h, supernatants were collected for cytokine measurement, and cells were harvested for analysis of ICAM-1 expression (data not shown). For each cytokine, data (n = 5–6) are plotted in nanograms per milliliter as means ± S.E.M.
Effect of PS-1145 and ML120B on NF-κB-Dependent Transcription in Primary HBE Cells. Collectively, the above data support the concept that small-molecule IKK inhibitors may elicit anti-inflammatory effects on the pulmonary epithelium. However, because A549 cells are a cancer-derived cell line, key aspects of the above study were confirmed in primary human airway epithelial cells. Therefore, normal primary HBE cells were cultured and then infected with an adenovirus carrying an NF-κB-dependent luciferase reporter construct. These cells were then pretreated with PS-1145 or ML120B before stimulation with IL-1β or TNFα (Fig. 7). Analysis of luciferase activity revealed a strong induction by either IL-1β or TNFα. The vehicle, DMSO, or the anti-inflammatory corticosteroid, dexamethasone, showed little or no effect on this induction. In contrast, PS-1145 and ML120B resulted in a significant concentration-dependent inhibition of both IL-1β-induced luciferase activity (EC50, 0.88 and 0.39 μM, respectively) and TNFα-induced luciferase activity (EC50 0.66 and 0.46 μM, respectively).
Effect of PS-1145 and ML120B on Inflammatory Gene Expression in Primary HBE Cells. To evaluate the effect of PS-1145 and ML120B on proinflammatory gene expression in primary HBE cells, ICAM-1 and IL-8 were selected as physiologically relevant target genes that may be robustly measured in small numbers of primary epithelial cells. Western blot analysis for ICAM-1 and GAPDH in the cellular extracts from the experiments presented in Fig. 7 revealed IL-1β- and TNFα-inducible expression of ICAM-1 (Fig. 8A). As with NF-κB-dependent transcription, there was little or no apparent effect of the vehicle, DMSO, or dexamethasone. However, both PS-1145 and ML120B produced consistent, and potent, concentration-dependent inhibitions of ICAM-1 expression. The efficacy of these effects matched closely the inhibition of NF-κB-dependent transcription observed in Fig. 7.

Effect of PS-1145, ML120B, and dexamethasone on NF-κB-dependent transcription in primary human airway epithelial cells. Primary human airway epithelial cells were infected with an adenoviral vector carrying an NF-κB-dependent luciferase reporter (Ad5-NF-κB-luc). After 24 h, the medium was changed to fresh supplement free media, and the cells were stimulated with IL-1β (A) (1 ng/ml) or TNFα (B) (10 ng/ml) in the presence or absence of a 1.5-h preincubation with 0.1% (v/v) DMSO, dexamethasone (1 μM) (left), or various concentrations of either PS-1145 or ML120B (right). After 8 h, cells were harvested for determination of luciferase activity. In each case, in A and B, data (left, n = 10; right, n = 5–7) are expressed as a percentage of stimulation and are plotted as means ± S.E.M.
Parallel analysis, by ELISA, of supernatants derived from the experiments depicted in Fig. 7, revealed robust increases in IL-8 release following both IL-1β and TNFα treatment (Fig. 8B) DMSO was without effect, and dexamethasone resulted in significant 30.8 and 26.1% reductions in IL-1β- and TNFα-induced expression of IL-8, respectively. Again, PS-1145 and ML120B produced concentration-dependent decreases in IL-1β-induced release, IL-8 release (EC50 1.6 and 23 μM, respectively), as well as TNFα-induced release of IL-8 (EC50, 4.8 and 5.7 μM, respectively) (Fig. 8C).
Although neither PS-1145 nor ML120B seemed to affect A549 cell viability, the possibility of effects on primary cell viability was also investigated (see supplemental data). Neither IL-1β nor TNFα in the presence or absence of PS-1145 or ML120B at concentrations of up to 30 μM revealed any significant effects on primary HBE cell viability over the time frames used in the above experiments (Supplemental Table 1; Supplemental Fig. 1).
Discussion
As noted above, the transcription factor NF-κB is considered a potential target for novel anti-inflammatory therapies in diseases such as asthma and COPD (Barnes and Karin, 1997; Karin et al., 2004). In our previous studies, we showed that overexpression of a dominant inhibitor of NF-κB or dominant-negative IKK2 prevented the activation of NF-κB and NF-κB-dependent transcription in pulmonary A549 cells in response to both IL-1β and TNFα (Catley et al., 2005). Since a parallel effect was observed on the expression of inflammatory genes, including ICAM-1, IL-8, and GM-CSF, and yet dominant-negative IKK1 was without effect, these data suggest that IKK2 is the dominant IKK in the activation of these genes. Furthermore, this suggests that inhibition of IKK2 may show anti-inflammatory potential. Given that IL-6 is a key inflammatory cytokine and is elevated in inflammatory diseases, including COPD (Chung, 2006), the current finding that the IL-1β- and TNFα-dependent induction of IL-6 is also both NF-κB- and IKK2-dependent lends considerable support for a putative anti-inflammatory effect of IKK2 inhibitors. Certainly, these findings are consistent with data from IKK1- and IKK2-deficient mice and suggest that IKK2 is the major IKK responsible for IL-1β- and TNFα-mediated induction of NF-κB activity (see Hayden and Ghosh, 2004). For small-molecule inhibitors of IKK2 to be effective in combating the inflammation associated with diseases such as asthma or COPD, it is imperative that these compounds are effective on the key structural cells of the airway. In this regard, IKK2-selective inhibitors have been shown to prevent inflammatory production of cytokines in human airway smooth muscle (Birrell et al., 2005; Catley et al., 2006; Issa et al., 2006). Indeed, an anti-inflammatory benefit for IKK2 inhibition is suggested in both lipopolysaccharide and allergen-sensitized in vivo models of lung inflammation (Birrell et al., 2005, 2006). In the current study, we extend this concept and show that inhibition of IKK2, using PS-1145 and ML120B (Hideshima et al., 2002; Castro et al., 2003; Nagashima et al., 2006; Wen et al., 2006), is highly effective at preventing both the activation of NF-κB-dependent transcription induced by IL-1β or TNFα, as well as the proinflammatory expression of ICAM-1 and the release of IL-6, IL-8, GM-CSF, RANTES, GROα, and MCP-1 in human pulmonary A549 cells. Furthermore, we confirm this ability of PS-1145 and ML120B to inhibit NF-κB-dependent transcription in primary HBE cells and show that the expression of ICAM-1 and IL-8 is also repressed. Given that NF-κBis activated in the airway epithelium of asthmatic patients (Hart et al., 1998), we believe our data strongly support the concept of using IKK2 inhibitors to target inflammatory gene expression by the epithelium. Furthermore, the proinflammatory and chemoattractant nature of the gene products examined in the current investigation is really just representative of the possible effects of an IKK2 inhibitor. For example, in addition to the impairment of ICAM-1 expression on the epithelium, it is highly likely that the expression, on both epithelial and other structural cells, of additional adhesion molecules and selectins, which are also NF-κB-dependent (Pahl, 1999), will also be impaired. This would further reduce recruitment of inflammatory cells to the airways. Likewise, in addition to IL-6, IL-8, GM-CSF, RANTES, GROα, and MCP-1, numerous other cytokines and chemokines are at least partly NF-κB-dependent, and the inhibition of these would have a profound collective effect on the airway inflammatory responses (Pahl, 1999). Given these data, it is likely that the recruitment of inflammatory cells to the airway epithelium would be severely impaired. Therefore, we predict that delivery of IKK2 inhibitors to the lung and, in particular, the airway epithelium will reduce proinflammatory responses.

Effect of PS-1145, ML120B, and dexamethasone on the expression of ICAM-1 and IL-8 in primary human airway epithelial cells. A, after the indicated treatments, cellular lysates from the experiments depicted in Fig. 7 were subjected to Western blot analysis for ICAM-1 and GAPDH. For each set of conditions (n = 5), blots were conducted, and a qualitatively similar result was obtained on each occasion. Representative examples are shown. B and C, supernatants from the experiments in Fig. 7 were harvested and analyzed for the presence of IL-8 after IL-1β or TNFα treatment. In each case, B (n = 10) shows absolute release of IL-8, whereas C (n = 5) shows the effect of PS-1145 and ML120B, and they are plotted as a percentage of IL-1β or TNFα-stimulated, respectively. All data are plotted as means ± S.E.M.
In the treatment of the airway disease, it is important that the integrity of the epithelium is maintained so that protective barrier function is not disrupted. In this context, we have previously noted that inhibition of NF-κB may promote apoptosis in A549 cells (Catley et al., 2005). Because apoptosis was also a feature in IKK2-deficient mice (Li et al., 1999; Tanaka et al., 1999), we tested the effects of both PS-1145 and ML120B on A549 and primary HBE cell viability. In each case, no changes in viability were detected over the experimental time frames used, suggesting that the observed inhibitory effects of PS-1145 and ML120B on inflammatory gene expression are not secondary to effects on cell viability. Although these data further support the possibility that IKK2 inhibitors may be of benefit in the context of airway inflammation, it is important to note that the current studies were conducted over relatively short time frames. Therefore, longer term analyses are necessary to fully exclude the possibility of any delayed effects of these compounds.
Corticosteroids remain the cornerstone of therapy for the vast majority of asthmatics (Barnes, 2001). However, in asthma, there a significant number of patients exist who remain poorly controlled by corticosteroids, and, in the case of COPD, corticosteroids are, at best, of modest benefit (Adcock and Ito, 2004; Barnes, 2004; Barnes and Hansel, 2004; Wouters, 2004). In A549 cells, we have previously documented 40 to 50% inhibition of NF-κB-dependent transcription by the corticosteroids dexamethasone and budesonide (Newton et al., 1998a; Chivers et al., 2004), whereas the expression of genes including COX-2, GM-CSF, or IL-8 were considerably more sensitive to repression by corticosteroids (Newton et al., 1998a; Chivers et al., 2004, 2006). Taken together, these data indicate that the targeting of NF-κB-dependent transcription by corticosteroids may represent a more minor mechanism of action than has been previously thought (Barnes and Karin, 1997; Barnes, 2001). In support of this statement, corticosteroid treatment failed to modulate activated NF-κB in the airways (Hart et al., 1998). Furthermore, we now report that although IL-8 expression was significantly repressed, the induction of NF-κB-dependent transcription in primary HBE cells was unaffected by a concentration of dexamethasone (1 μM) that produces near-maximal inhibitory effects in both A549 and primary HBE cells (Kwon et al., 1994; Newton et al., 1998b; Chivers et al., 2006). More importantly, in the study of Kwon et al. (1994), dexamethasone-dependent repression of IL-8 in primary epithelial cells revealed a very similar overall level of repression to that observed in the current study, and this effect occurred with an EC50 in the region of 10 nM. We therefore believe that, rather than requiring higher concentrations of corticosteroid or longer incubation times, these results reflect the true repressive ability of dexamethasone in HBE cells. Consistent with this, there was no noticeable effect of corticosteroid on the expression of ICAM-1, whereas inhibition of IKK and NF-κB resulted in a profound repression of ICAM-1 expression. Thus, IKK2 inhibitors seem to target separate processes to corticosteroids in the repression of inflammatory gene expression. This finding may have a considerable impact in the context of steroid insensitive-asthma, COPD, and in exacerbations of both asthma and COPD, which are often poorly responsive to corticosteroid therapy (Adcock and Ito, 2004; Barnes, 2004; Barnes and Hansel, 2004; Wouters, 2004; Proud and Chow, 2006). Thus, in disease exacerbations, in which corticosteroids are already being administered, the addition of an IKK2 inhibitor may provide considerable additional anti-inflammatory benefit by enhancing both the repression of inflammatory genes that are already partially targeted by corticosteroids as well as by repressing genes that were not steroid-responsive.
In summary, we demonstrate that targeting IKK2 with small-molecule inhibitors is effective at preventing the activation of NF-κB in pulmonary epithelial cells. Furthermore, this manipulation results in the reduced expression of proinflammatory genes in both A549 cells and primary HBE cells. Based on the inflammatory roles of genes such as ICAM-1, IL-8, IL-6, GM-CSF, RANTES, GROα, and MCP-1 and the key role of the airway epithelium in the pathogenesis of asthma and COPD, we predict that inhibitors of IKK2 may represent valuable additional anti-inflammatory agents in management of these diseases. It is noteworthy that our data suggest that IKK inhibition may be at least partially complementary to the effects of corticosteroids. Therefore, IKK2 inhibitors or other inhibitors of NF-κB may provide anti-inflammatory benefit in exacerbations of asthma or COPD or in other inflammatory airway diseases, including cystic fibrosis, where corticosteroids are of limited benefit.
Footnotes
-
This study was supported by a grant from Millennium Pharmaceuticals to Imperial College London. R.N. is a Canadian Institutes of Health Research New Investigator and an Alberta Heritage Foundation for Medical Research Scholar.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.118125.
-
ABBREVIATIONS: COPD, chronic obstructive pulmonary disease; IL, interleukin; TNF, tumor necrosis factor; RANTES, regulated and activation normal T cell expressed and secreted; MCP, monocyte chemotactic protein; GRO, growth-related oncogene; ICAM, intercellular adhesion molecule; NF, nuclear factor; IKK, IκB kinase; HBE, human bronchial epithelial; PS-1145, N-(6-chloro-9H-β-carbolin-8-ly) nicotinamide; ML120B, N-(6-chloro-7-methoxy-9H-β-carbolin-8-yl)-2-methyl-nicotinamide; DMSO, dimethyl sulfoxide; MOI, multiplicity of infection; EMSA, electrophoretic mobility shift assay; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ELISA, enzyme-linked immunosorbent assay; GM-CSF, granulocyte macrophage-colony-stimulating factor.
-
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material. - Received December 3, 2006.
- Accepted February 20, 2007.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}